Continuing on the information that David Sinclair provides in his book, we’ll have a look here at the 3 pathways he mentioned. Sirtuins, AMPK and mTOR and how these are influenced with a ketogenic diet.
mTOR
According to his book, mTOR should be kept at low activity. Whenever mTOR is stimulated then the repair mechanisms can’t take be active. There are 2 main causes for activated mTOR and those are insulin and protein. So taking out carbs and protein from our food sets us up for longevity! Not so fast. While you can take out carbs completely, you do need protein. Don’t just think of protein as muscle meat. Every single cell in your body is made up and contains if not thousands of molecules made up from the individual amino acids that make up protein. These proteins have functions such as enzymes to make things happen in your body. There is a minimum that you need just to live.
A ketogenic diet is ketogenic simply because it restricts carbohydrates. It is a condition to produce ketones in your body. The protein side is a bit more grey zone in the amount that you can eat for ketogenesis but by increasing your fat intake you can limit your protein intake naturally as the fat will satiate you.
Eating zero carbohydrates, high in fat and not-more-than-sufficient protein will minimize mTOR and increase ketone production. Sorry to be so vague on the protein but in the science field numbers range wildely and there are no decent conclusions long term.
Personally I choose to aim for 1gr of protein per lean kg of body mass (total weight – fat weight).
Anyway, a reduced mTORC1 activity is needed in the liver to allow for ketone production so ketones can be taken as evidence of reduced mTOR.
The mechanism for the effect on cardiac hypertrophy appears to be inhibition of HDACs that suppress the activity of a mechanistic mTOR complex (88). This is one of several examples of intersections between BHB, its signaling effects, and mTOR/rapamycin, a canonical longevity-regulating pathway (55). As described above, mTOR is also a checkpoint in the activation of ketogenesis; inhibition of mTORC1 is required to activate the transcription factors and hormones that control ketogenesis (4, 117).
“β-Hydroxybutyrate – A Signaling Metabolite”, John C. Newman and Eric Verdin, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6640868/
Wait a minute! I’ve read that ketones increase mTOR! That is correct. The ketone body beta-hydroxybutyrate (BHB) has different effects. Keep in mind that the study referenced below is about a ketone ester supplement (so not an endogenous increment) but nonetheless, and that it shows here a synergistic effect during protein intake AND the effect is noted in skeletal muscle where you want mTOR to be up temporarily in order to build muscle. It could be that this is happening in other organs as well.
Under normal circumstances insulin gets activated when eating. Only a very modest amount on a ketogenic diet but potentially enough to bring down blood BHB levels to lower than 1 or even 0.5. It would be a great experiment to see how fast BHB levels go up after feeding, what the plasma amino acids are across this time and how it affects muscle protein synthesis.
“Intake of a Ketone Ester Drink during Recovery from Exercise Promotes mTORC1 Signaling but Not Glycogen Resynthesis in Human Muscle”, Tijs Vandoorne, Stefan De Smet, Monique Ramaekers, Ruud Van Thienen, Katrien De Bock, Kieran Clarke, and Peter Hespel, 2017, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5440563/
“Effect of beta-hydroxybutyrate on whole-body leucine kinetics and fractional mixed skeletal muscle protein synthesis in humans.”, K S Nair, S L Welle, D Halliday, and R G Campbell, 1988, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC303494/
I’m of the opinion that longevity by restricting protein works but only if this is done while having elevated ketone levels in the blood. You need to protect those muscles from breaking down and preferably even build them up.
“Role of Dietary Protein and Muscular Fitness on Longevity and Aging”, Barbara Strasser, Konstantinos Volaklis, Dietmar Fuchs, and Martin Burtscher, 2018, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5772850/
This makes the ketogenic diet an ideal fit. Low mTOR while fasted, extra driver of muscle protein synthesis when eating protein. Ideally after exercise when those circulating fatty acids still generate increased ketones.
Since one of the studies was about exercise… shortly after exercise your endogenous ketone production goes up and then gradually settles down again to baseline levels. So dietary protein intake is likely optimal shortly after exercise when your ketones are up highest. Do check out different types of exercise to see what it does to ketone levels though.
“Post-exercise ketosis”, J H Koeslag, T D Noakes, and A W Sloan, 1980, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1279383/
“POST-EXERCISE KETOSIS”, R.H. Johnson, J.L. Walton, H.A. Krebs, D.H. Williamson, 1969, https://www.thelancet.com/journals/lancet/article/PIIS0140673669909313/fulltext
Not only do we require low mTOR to produce ketones, ketones themselves lower mTOR. This has been found in the brain in animal experiments so caution to extrapolate this to human results. It will be hard to prove since we can’t take a sample from a living person 😦 But the pathway is described and for now I don’t see any reason this would not be the case for us as well.
“The ketogenic diet inhibits the mammalian target of rapamycin (mTOR) pathway”, McDaniel SS, Rensing NR, Thio LL, Yamada KA, Wong M, 2011, https://www.ncbi.nlm.nih.gov/pubmed/21371020/
AMPK
We see that AMPK is elevated on a ketogenic diet. These studies are done on rodents which kind of makes it hard to accept as evidence. The reason for this is that these buggers have a high metabolism, around 7-fold higher than humans. For this reason their protein intake has to be severely restricted or they can’t get into ketosis. They’ll turn much of the protein into glucose otherwise.
“A high-fat, ketogenic diet induces a unique metabolic state in mice.”, Kennedy AR, Pissios P, Otu H, Roberson R, Xue B, Asakura K, Furukawa N, Marino FE, Liu FF, Kahn BB, Libermann TA, Maratos-Flier E, 2007, https://www.ncbi.nlm.nih.gov/pubmed/17299079/
However, the above publication also refers to other research showing that AMPK is upregulated when glucose metabolism goes down. That makes it more likely to happen in humans as well.
AMPK responds to ATP depletion and is activated by low glucose (27); AMPK may also be inhibited by insulin in some cases (50). Activation of AMPK leads to decreased fatty acid synthesis and increased fatty acid oxidation (20, 27)
Despite being another animal study, the researchers also decided to test via cell culture if ketones directly affect AMPK and found it to be the case.
“β-Hydroxybutyrate suppresses inflammasome formation by ameliorating endoplasmic reticulum stress via AMPK activation”, Ha Ram Bae, Dae Hyun Kim, Min Hi Park, Bonggi Lee, Min Jo Kim, Eun Kyeong Lee, Ki Wung Chung, Seong Min Kim, Dong Soon Im, and Hae Young Chung, 2016, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5341812/
So it seems the we can safely assume in humans that AMPK goes up on a ketogenic diet. The diet keeps insulin low, there’s the ketones that induce AMPK and BHB also suppresses hepatic glucose output so there is a lower glucose level. BHB suppressing hepatic glucose output is not referenced here but this is visible in the human studies where they administer exogenous ketones. It is also a suppressive effect that must take place or you end up in ketoacidosis thanks to the CO2 contribution of metabolizing glucose.

“Ketosis, ketogenic diet and food intake control: a complex relationship.”, Paoli A, Bosco G, Camporesi EM, Mangar D, 2015, https://www.ncbi.nlm.nih.gov/pubmed/25698989
Sirtuins
There are different sirtuins, 1 to 7, likely involved in longevity but SIRT1 is known to be directly involved in DNA repair so we’ll focus on that one. As explained in the book, these sirtuins require NAD+ to function. This gives ketones 2 possible ways to activate sirtuins. Either somewhat directly or via NAD+ production or saving.
Cockayne syndrome is a disease with accelerated aging caused through increased DNA repair. Similar to how the book describes they were able to accelerate aging in mice by deliberately breaking DNA. The next study also comments on how NAD+-dependent SIRT1 is required for this DNA repair. The disease causes higher activation of PARP1 which is also dependent on NAD+ resulting in a lower availability for SIRT1.
BHB caused an increase in SIRT1 activity. That in itself is encouraging but is it because it reduced PARP1 activity? Did it cause more NAD+ production? Did it directly stimulate SIRT1, bypassing the need for NAD+? Lots of questions we’d like to know an answer to. We want to make sure it is not an effect that is specific to Cockayne syndrome. They did experiment with PARP inhibition, leading to higher SIRT1 activity as it preserved NAD+ so this is another angle we can look at for BHB.
They did a lot of investigation and found BHB in cell culture would elevate acetyl-coa availability which regulate histone acetylase activity. So SIRT1 activity was increased indirectly by BHB but SIRT1 is a histone deacetylase.
“A high-fat diet and NAD(+) activate Sirt1 to rescue premature aging in cockayne syndrome.”, Scheibye-Knudsen M, Mitchell SJ, Fang EF, Iyama T, Ward T, Wang J, Dunn CA, Singh N, Veith S, Hasan-Olive MM, Mangerich A, Wilson MA, Mattson MP, Bergersen LH, Cogger VC, Warren A, Le Couteur DG, Moaddel R, Wilson DM 3rd, Croteau DL, de Cabo R, Bohr VA, 2014, https://www.ncbi.nlm.nih.gov/pubmed/25440059
This needs to be explained a bit further. BHB is a histone deacetylase (HDAC) inhibitor. Sirtuins (1-7) belong to the class III HDAC’s while BHB inhibits class I & II HDAC’s. I was first thinking that, by inhibiting activity of other HDAC’s that BHB saves NAD+ consumption, leaving more available for the sirtuins. But those class I & II HDAC’s are zinc dependent rather than NAD+. So they do not oppose each other, neither does it help save NAD+.
“Suppression of Oxidative Stress by β-Hydroxybutyrate, an Endogenous Histone Deacetylase Inhibitor”, Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, Grueter CA, Lim H, Saunders LR, Stevens RD, Newgard CB, Farese RV Jr, de Cabo R, Ulrich S, Akassoglou K, Verdin E, 2013, https://www.ncbi.nlm.nih.gov/pubmed/23223453
So far it seems BHB increases histone acetylation through increased availability of acetyl-coa and causes increased expression of FOXO3A and MT2 (explained in the above paper) through the specific HDAC inhibition. Somehow this leads to increased SIRT1 activity.
As it turns out, it is FOXO3A that is required for SIRT1 expression (in combination with p53).
“Nutrient availability regulates SIRT1 through a forkhead-dependent pathway.”, Nemoto S, Fergusson MM, Finkel T, 2004, https://www.ncbi.nlm.nih.gov/pubmed/15604409
This is really great because it shows the indirect effect BHB has on SIRT1 expression via FOXO3A thus a mechanism that is independent of our Cockayne syndrome.
NAD+
As mentioned at the beginning, sirtuins are dependent on NAD+. As it turns out, Acetoacetate (AcAc) is the first metabolite in the pathway to BHB. In order to produce BHB, AcAc has to combine with NADH and H+. The result is BHB and NAD+. So at least in the liver there is extra support for sirtuin activity.
https://en.wikipedia.org/wiki/3-Hydroxybutyrate_dehydrogenase
We also have evidence of ketone body production in astrocytes. Particularly in the ventromedial hypothalamus so at least that part of the brain could benefit from the addition in NAD+.
“Fatty acid-induced astrocyte ketone production and the control of food intake”, Christelle Le Foll and Barry E. Levin, 2016, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4935491/
We could set our hope on a surplus in NAD+ availability, hopefully exported into the bloodstream and distributed across the body. There is a potential for it via the Connexin 43 transporter.
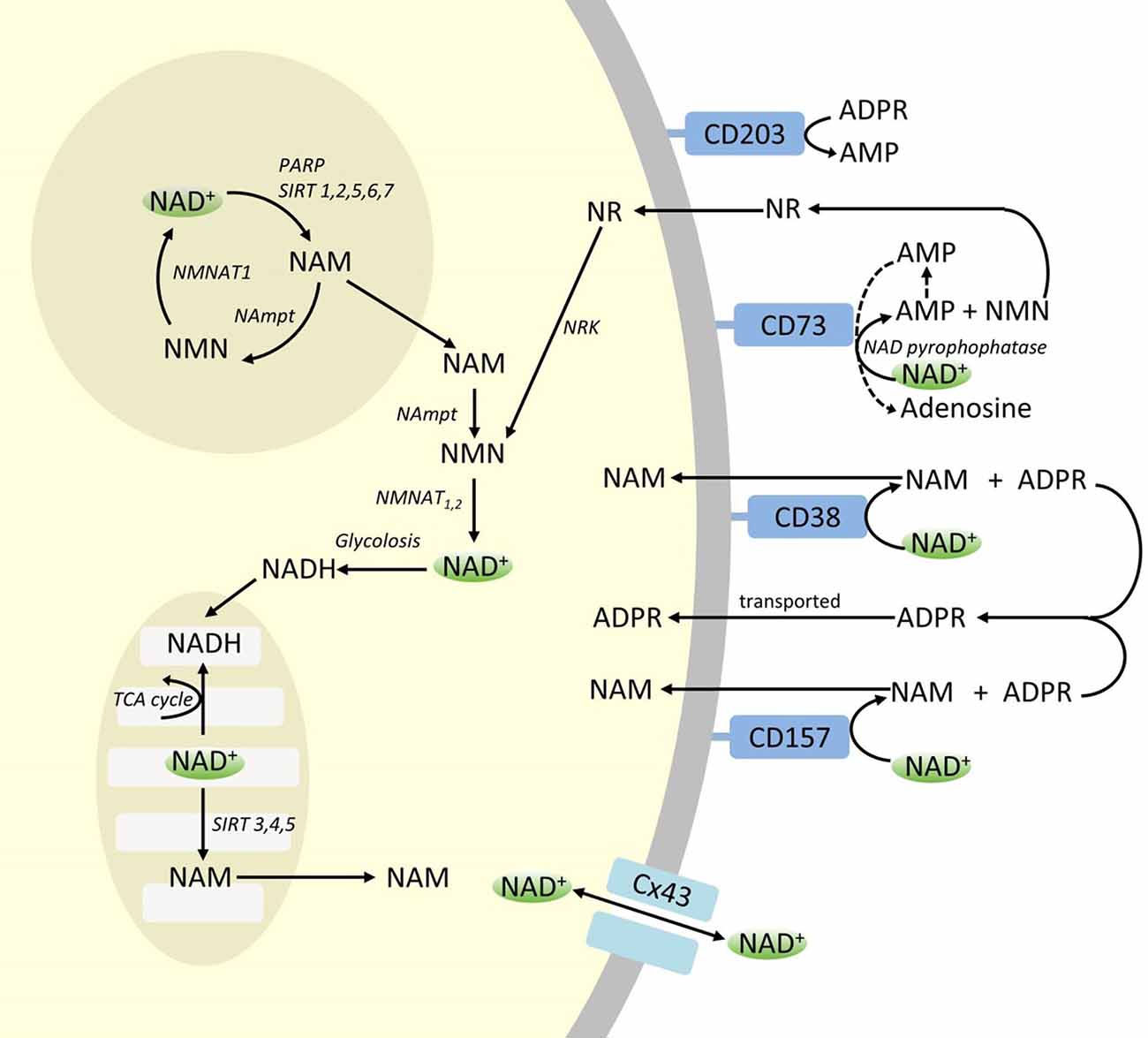
“A Pilot Study Investigating Changes in the Human Plasma and Urine NAD+ Metabolome During a 6 Hour Intravenous Infusion of NAD+”, Ross Grant, Jade Berg, Richard Mestayer, Nady Braidy, James Bennett, Susan Broom and James Watson, 2019, https://www.frontiersin.org/articles/10.3389/fnagi.2019.00257/full
“Exogenous nicotinamide adenine dinucleotide regulates energy metabolism via hypothalamic connexin 43”, Eun Roh, Jae Woo Park, Gil Myoung Kang, Chan Hee Lee, Hong Dugu, So Young Gil, Do Kyeong Song, Hyo Jin Kim, Gi Hoon Son, Rina Yue, Min-Seon Kim, 2018, https://www.sciencedirect.com/science/article/abs/pii/S0026049518301732
Update: 2020-01-01
As mentioned above, BHB production leads to the generation of NAD+ but BHB conversion to acetyl-Coa consumes NAD+ because the conversion back to AcAc requires this before it can be converted to acetyl-Coa.
But this is still far better than glucose metabolism which utilizes 2 NAD+ molecules per acetyl-Coa generated while dietary glucose doesn’t create any NAD+.
In addition AcAc also circulates in the body. If it gets used directly for energy so skipping the conversion to BHB then it doesn’t consume NAD+ when converting to acetyl-coa. This means ketone metabolism is a neutral effect on NAD+. Although, by replacing glucose metabolism, it preserves NAD+ resulting in a higher NAD+/NADH ratio.

“Ketone-Based Metabolic Therapy: Is Increased NAD+ a Primary Mechanism?” – “Mechanisms of Ketogenic Therapy: Evidence for Increased NAD+”, Marwa Elamin, David N. Ruskin, Susan A. Masino, and Paola, Sacchetti, 2017, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5694488/#s2title
Finally
We found evidence that the ketogenic diet works on all 3 levels expressed in the book of David Sinclair. This is very encouraging.
What still remains open is how well we can maintain ketosis. And if we can generate sufficient ketones, do they, and the NAD+, end up in every cell of our body or are there only specific organs affected? It is easier for rats in a lab to be in lifelong ketosis but humans don’t live there and are not rats.
At least the available data makes me hopeful and so do all animal experiments where they show the life extending or at least health-span extending abilities of the ketogenic diet.
— The End —

Leave a comment