Perhaps not a very sexy name but why name it something else than what it is?
After studying the functioning of our lipoproteins for a good 2 years (and still not finished learning!), it has finally become clear what those lipoprotein are meant for. As complex as it may be, the end goal is really simple:
- To facilitate the storage of fat
- To prepare for the storage of fat
- To recuperate storage structure when it is no longer needed
- Transfer fat storage from one location to another
Compare it with the complexity of a swiss watch. All that mind-boggling complexity of the internal components, in the end to simply show you the hour, minutes and seconds.
I want to show you how these lipids relate to the storage objective. Not only that, by understanding how it functions I also want to provide a perspective on how the system can be protective of atherosclerosis, indeed even reverse plaque formation (!)
A very bold claim to make indeed. Just consider it speculative from my side. I present you the info and you make up your own mind. It will never be substantial until research is done to elucidate this further.
What I’m going to present you now, although elaborate, is still a simplified picture. Elaborate enough to show how the system works and simplified enough to show the most important components.
The basics
This picture, for most people who have already looked into the lipids, will be fairly familiar.
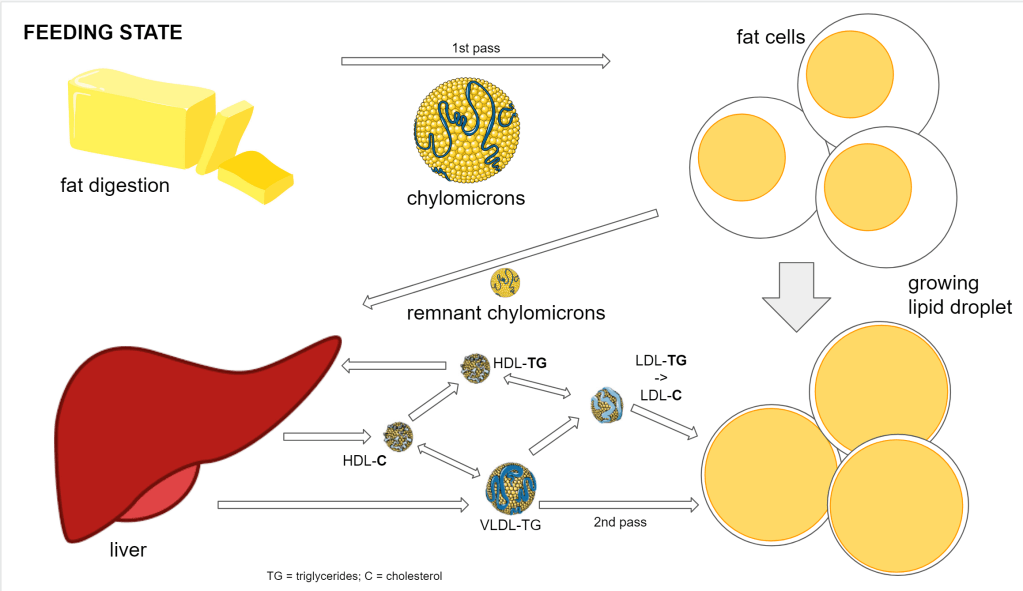
- Dietary fat gets stored in adipose tissue
- 1st phase delivery is done by chylomicrons
- 2nd phase delivery is done by the interplay of various lipoprotein (VLDL, IDL, LDL, HDL)
The role of liver-produced lipids and cholesterol
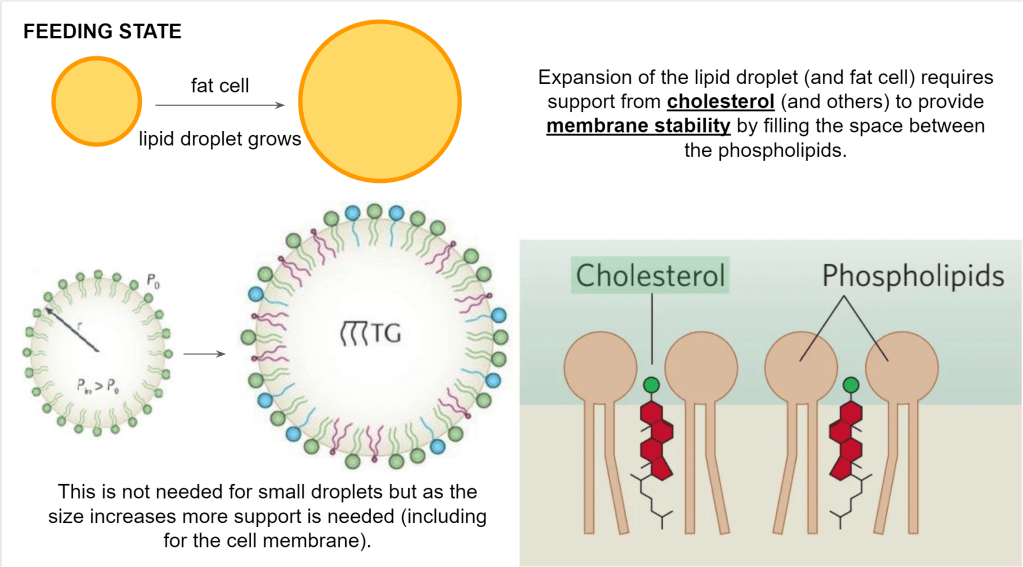
The fat needs to be pushed in a lipid droplet which resides inside the fat cell. So both the lipid droplet and the fat cell need to expand. They both have a membrane so the membrane needs to expand as well and that requires phospholipids and cholesterol.
Those phospholipids and cholesterol come from the liver-produced lipoprotein (VLDL, IDL, LDL). Keep those phospholipids in mind when you see growing or shrinking lipid storage because I won’t mention them any further.
Not important for the system although interesting, the type of fatty acids used to make up the phospholipids will determine how much stability is required and thus how much cholesterol will be absorbed in the membrane. This is why cholesterol values can vary depending on the type of fat you eat.
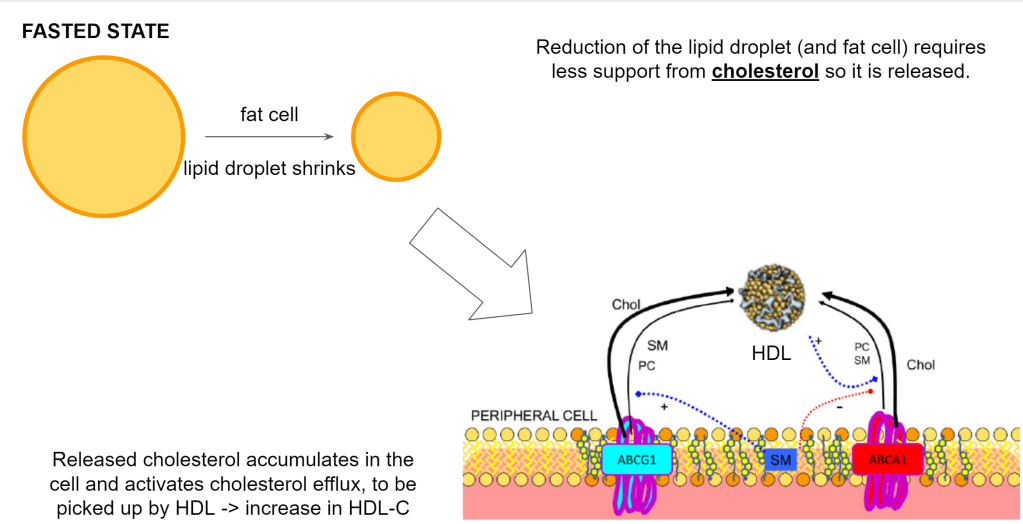
When we get into the fasted state, the fat cells will start to release the fat. Now the reverse happens. Both the lipid droplet and the fat cell will shrink so they need to reduce their membranes and thus also release the cholesterol that is in it.
This means the cell will have too much cholesterol and will get rid of it via HDL.
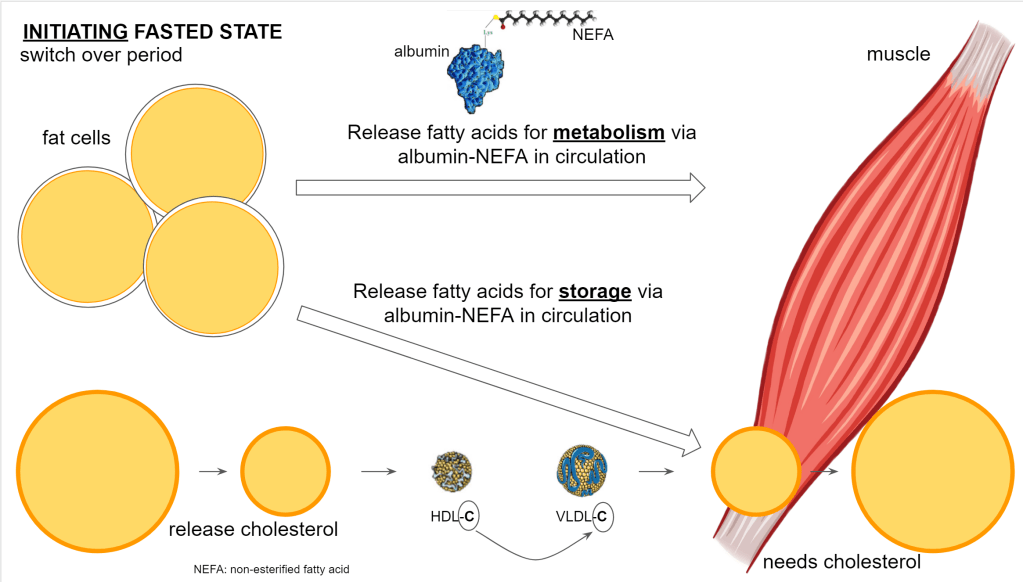
The fat goes out of the fat cells and will be used throughout the body. In the muscle cells specifically, the cells will store the lipids that may have been reduced from previous activity. So here the lipid droplets will grow and, you guessed it, also here the membrane of the droplet requires cholesterol.
At the bottom left you see the shrinking lipid content represented of the fat cells while on the bottom right you have the growing lipid content of the muscle cells.
As we proceed longer into the fasted state, the muscle reaches an equilibrium where the size of the lipid droplet is maintained at a roughly equal size. Keep in mind though, there is a high turnover in the lipid droplet. So there is a continuous breakdown and buildup of triglycerides but the end result is essentially zero change in size. There is no need for additional cholesterol or the removal of cholesterol.
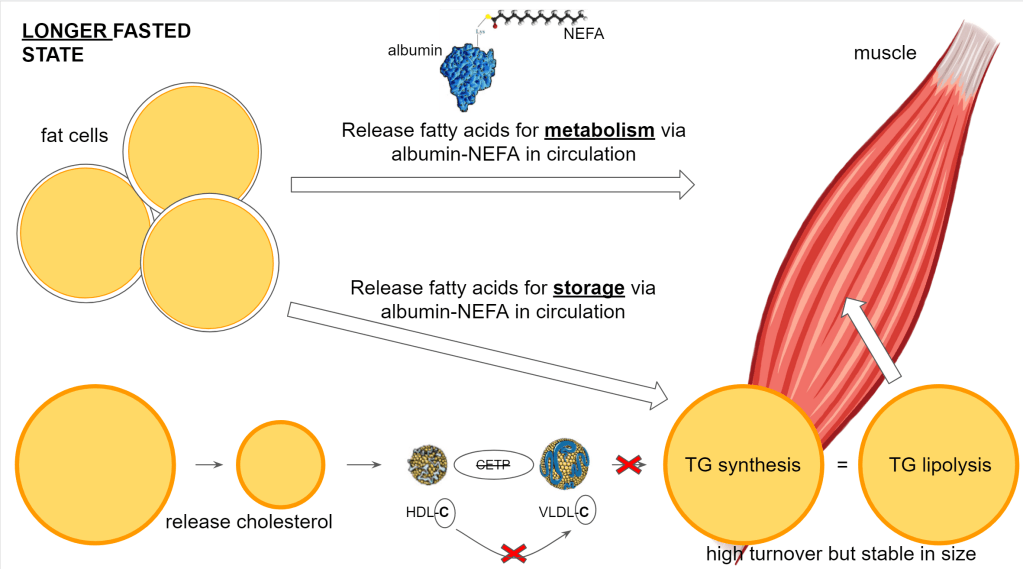
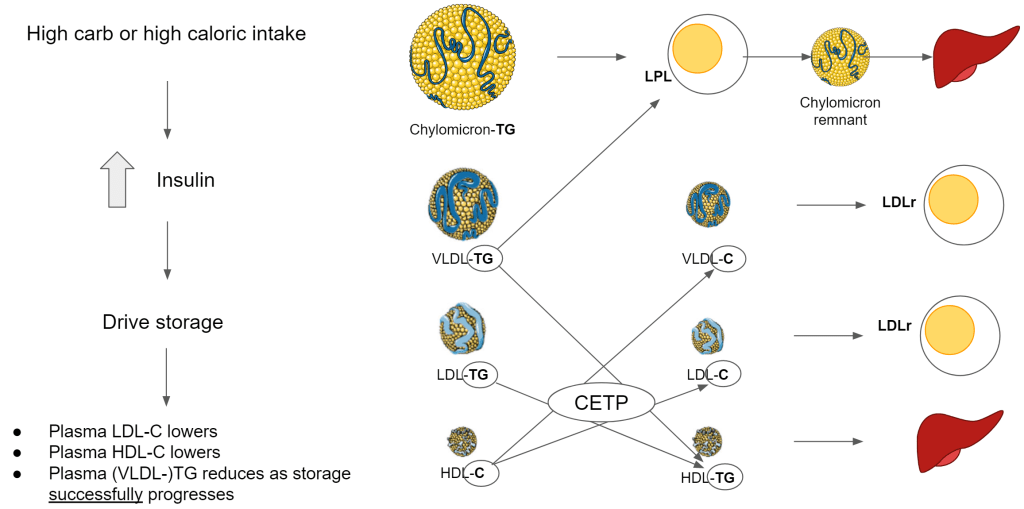
Before the lipoprotein can support this delivery of membrane components, they must have the right composition. This change in composition is facilitated by cholesterol ester transfer protein (CETP). The liver produces CETP under stimulation of insulin so the level of insulin drives the level of CETP activity.
With CETP:
- HDL-C exchanges cholesterol for triglycerides (TG) with VLDL, IDL and LDL (ApoB100 containing lipoprotein)
- HDL is dedicated to remodeling ApoB100 lipoprotein
- This means that VLDL, IDL and LDL lose their TG and in return gain cholesterol. They become cholesterol-enriched.
- This also means that HDL loses its cholesterol content and increases its TG content.
- By being TG-rich, more HDL will be taken up by the liver creating lower circulating HDL particles in the blood
- By being cholesterol-enriched, ApoB100 lipoprotein can be taken up by the cells
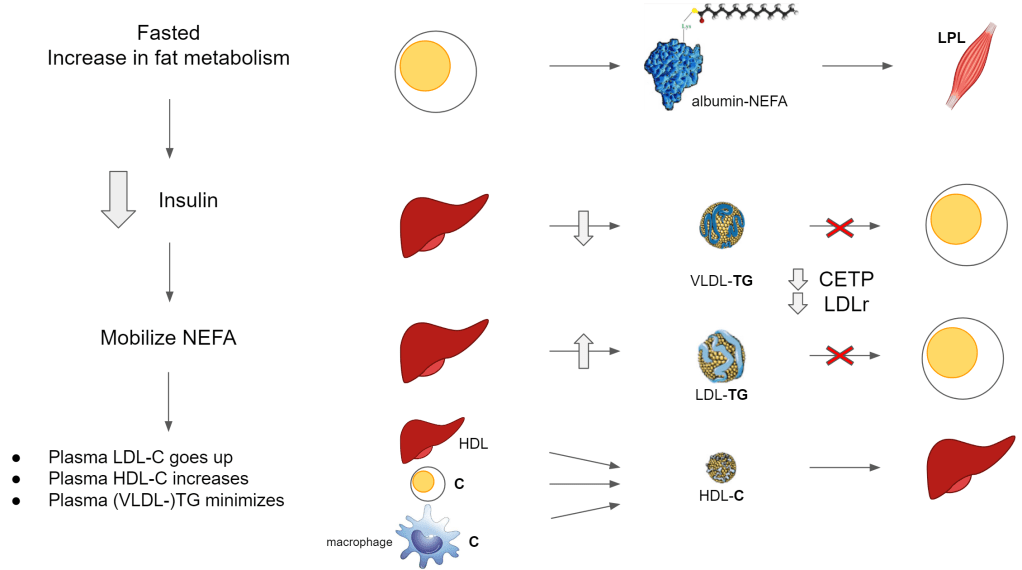
Without CETP:
- HDL cannot offload its cholesterol to the ApoB100 lipoprotein and instead stays in circulation longer and accumulates more cholesterol
- HDL is dedicated to cholesterol collection from cells
- ApoE and LCAT drive HDL cholesterol accumulation so that it becomes big enough for uptake by the liver
- The ApoB100 lipoprotein remain rich in TG and cannot be taken up by the cells
This is already becoming complex but you get a picture of how important CETP is. CETP will come back when we look at atherosclerosis..
Also note in the illustrations above, the free fatty acids aka non-esterified fatty acids (NEFA) (meaning not bound their usual glycerol backbone) are transported via albumin. This is an abundant available protein responsible for transporting the energy itself that will be used directly in the cells and also will be esterified (binding to glycerol) as triglycerides and stored in the lipid droplets inside the cells.
Getting everything across the cell membrane
There are 2 important factors to absorb the fatty acids and membrane structures. Note again, we focus primarily on cholesterol although the phospholipids are part of it.
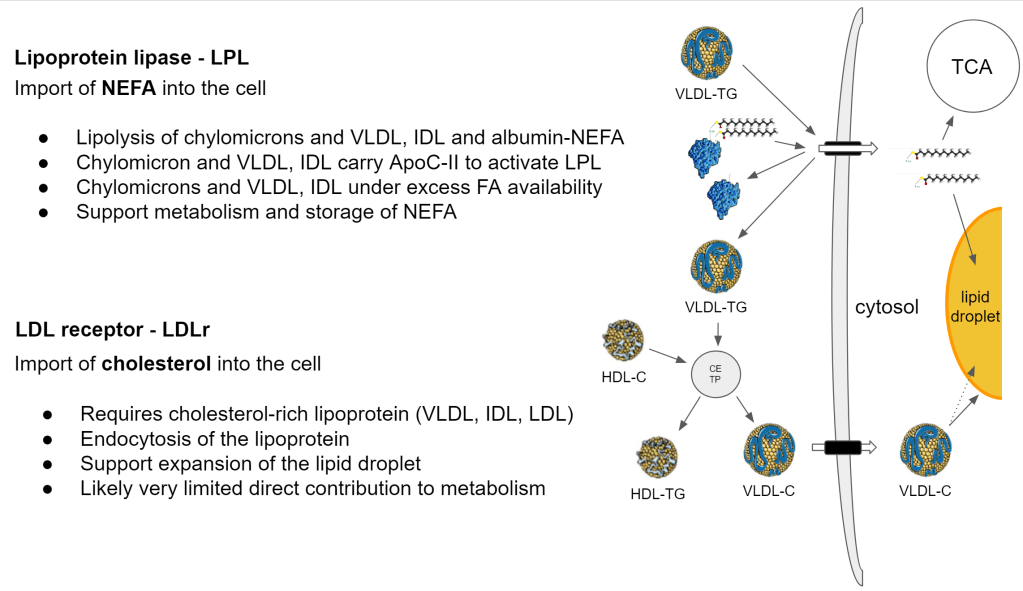
These are not just gates that are open 100%, accepting anything at all times. How are they regulated? How do we get the NEFA and cholesterol across?
LPL
- ApoC-II reduces on a high fat diet. This means that more NEFA is imported via albumin
- Sterol regulatory element-binding protein 1 (SREBP1) is activated under lower cellular cholesterol content and reduces LPL expression
- => This is a negative feedback which reduces NEFA import. When there is a shortage of cholesterol to build storage then you need to lower NEFA to avoid accumulation in the cell
- The fat cell LPL is activated by insulin. Muscle cell LPL is activated by low insulin
- => Insulin prioritizes fat storage in fat cells. => When insulin lowers, the energy is distributed and taken up by the other cells
- LPL is increased during and after exercise. During those moments, there is an increased demand for energy
LDLr
- Requires cholesterol-rich lipoprotein (VLDL, IDL, LDL)
- Endocytosis of the lipoprotein (complete uptake of the lipoprotein)
- Support expansion of the lipid droplet membrane via cholesterol import
- Insulin activates SREBP1 and SREBP1 activates LDLr. As expected because SREBP1 shows there is a need for cholesterol and when driven by insulin it means we are focusing on storing fat.
LDLr in the muscle
- exercise, fasting activates SREBP1 via MAPK. Similar to insulin, we also want to store fat simply to respond to a higher fat metabolism need. When fat metabolism increases, there is need for a bigger local buffer, a bigger lipid droplet.
In summary so far, we can say that a diet existing out of high fat quantities in combination with a high fat metabolism causes a continuous growing and shrinking of the fat cells. If that is combined with exercise then also the muscles are faced with this growing and shrinking. The timeframe and the volume of fat handled causes those cells and lipid droplets to grow to larger sizes and to shrink to smaller sizes, compared to a diet and lifestyle where glucose is a major energy source. This leads to a lower flux of the fat storage.
Such an intensified remodeling on high fat/high activity requires more support from the circulating lipoprotein!
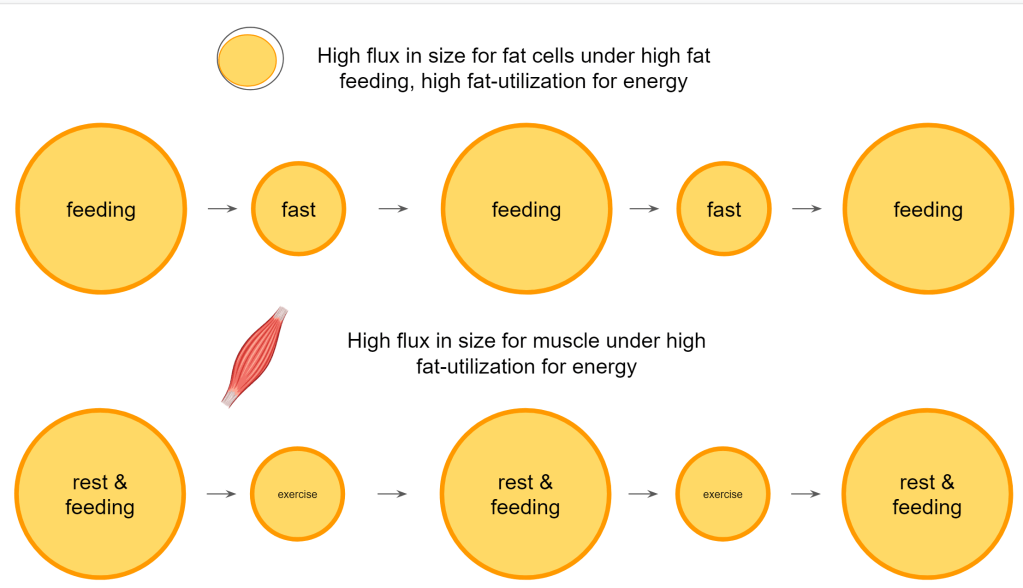
This has to be supported by the lipoproteins that we are familiar with. VLDL, LDL and HDL. Moreover, in order to be able to receive the next bolus of incoming fat it is good to have the right types and level of lipoprotein ready to support this.
What is the best way to accommodate a high fat meal so that we can swiftly provide the necessary membrane components for expansion?
- TG-rich (large buoyant) LDL
- High cholesterol levels on HDL
When the meal comes in, insulin comes up and will stimulate CETP. With those 2 listed above, CETP can immediately remodel LDL to become enriched with cholesterol so that it can be taken up by the fat cells. Both lipoproteins need to be sufficiently high to support a faster clearance of the fat into storage.
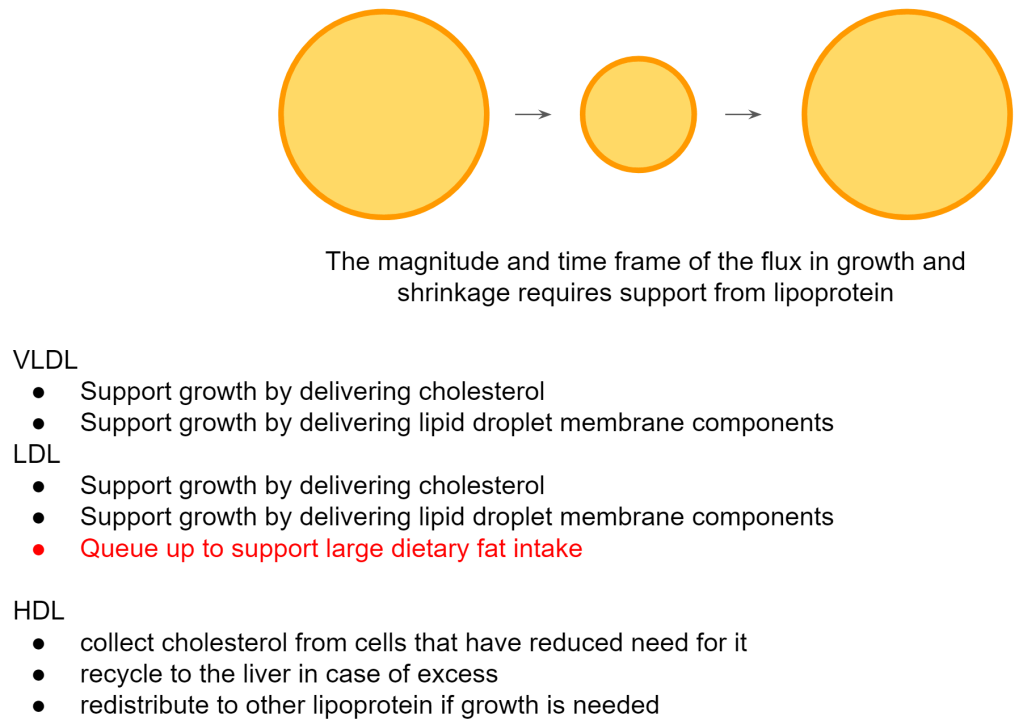
It’s my engineer thinking but would it be just a coincidence that LDL goes up under such high fat intake and consumption? This high flux signals the need for a greater storage capacity so doesn’t it make sense to have more of the membrane components ready to maximize the storage capacity? Could that be the reason why fasting LDL particles and HDL cholesterol are high? As soon as CETP kicks into action you’ll now have a lot of LDL particles ready to accept cholesterol and support the membrane expansion of fat cells and their lipid droplets.
High fasting LDL, HDL and low TG for lean individuals on a ketogenic diet
With the above information you understand the need for it. But how do we arrive at that situation? The following are general mechanisms that apply to everybody but the extend to which they get applied is determined by many factors such as the amount of fat you eat, exercise level, your level of insulin etc… There are other factors but insulin is a very big player in all of this.
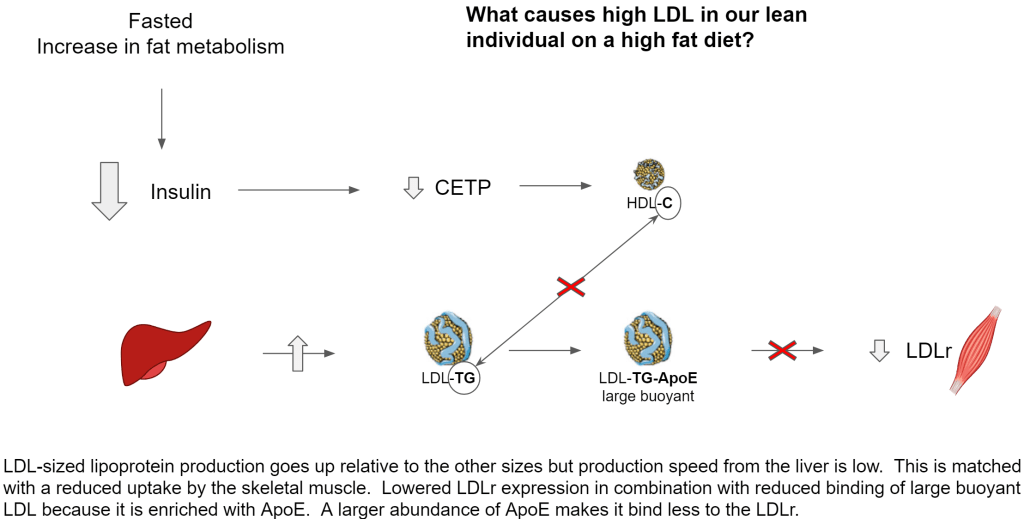
- Under fasted conditions, insulin is sufficiently low to keep cholesterol production very low. When fasted we are not interested in storing fat. The liver preserves most fatty acids for ketogenesis, bile production and its own fat metabolism. Both fatty acids and cholesterol production is low in such a way that it has shifted from VLDL production towards large buoyant LDL
- Large buoyant LDL stays in circulation longer because it is cholesterol poor, ApoE rich and thus has a lower ability to be taken up by LDLr for endocytosis. That ApoE enrichment is also the reason why small dense LDL (sdLDL) has a lower affinity for LDLr
- Low insulin keeps CETP low, thus VLDL and LDL cannot acquire sufficient cholesterol from HDL for endocytosis by LDLr
- Due to low CETP, the cholesterol load on HDL increases and minimizes exchange with VLDL, IDL, LDL.
- With very low to no production of VLDL sized lipoprotein, there are almost no lipoproteins that can acquire ApoC-II for lipolysis by LPL thus almost all skeletal muscle NEFA are derived from albumin
So on the production side we have lower production but more of the lipoprotein in the LDL size, in circulation there is no remodeling taking place and due to this the uptake by the skeletal muscle is much lower.
When the skeletal muscles are full and reduce their uptake of fatty acids, this will lead to a higher return of those fatty acids towards the liver. So the release of fatty acids from the fat cells is temporarily higher than the uptake by the other cells. This accumulation leads to a larger availability of fatty acids in the liver so that it can augment its production of TG-rich LDL.
Now it will depend on how big this discrepancy is. This higher level of fatty acids in the liver also causes more ketones (BHB) to be produced. The body may respond to this by slightly increasing insulin. This will cap BHB production, reduce the rate of fatty acid release from fat cells and slightly increase cholesterol production allowing the liver to temporarily produce more VLDL-sized lipoprotein. It will also slightly elevate CETP so that storage can be facilitated to get rid of the excess fat in circulation.
You may notice this as a slight elevation in your fasted triglycerides. It is only temporarily as it is part of an exercise to balance out demand and supply.
Buffering for energy usage
and issues with it
Still following so far? OK. Now lets have a look at issues with storage.
Just like with glucose, the fat is stored in large quantity in a central place and from there distributed to local storage in the muscle. Central is put in quotes, mainly referring to the adipocytes. It is supposed to sit under our skin and well.. our skin is completely wrapped around our body so it is a bit strange to say central.
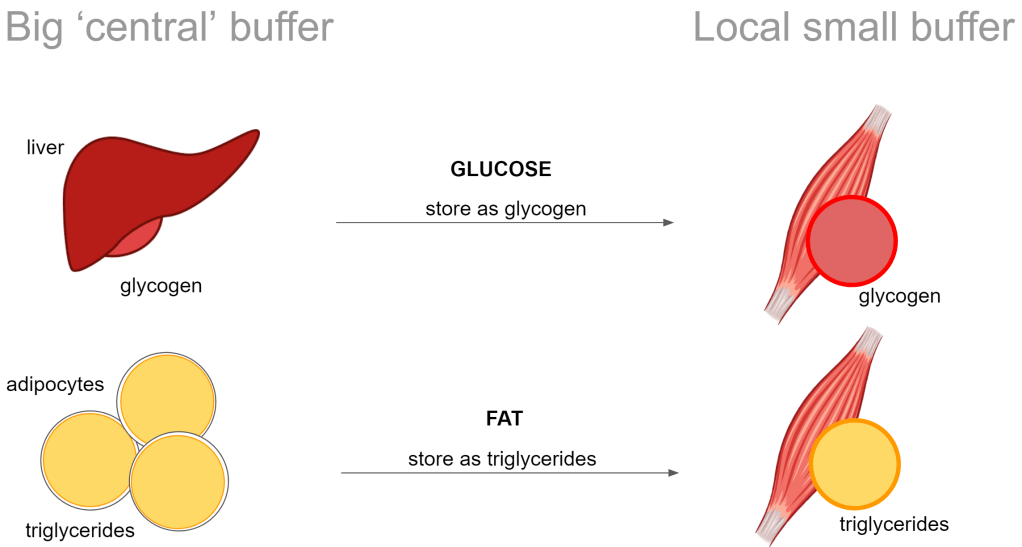
I’ve illustrated glycogen here just for comparison. Just note that I’m continuing only talking about the fat.
Storage limitation -> muscle insulin resistance
At some point, all of these buffers have to signal when they are full and (temporarily) cannot take in any more fat. This signaling is provided by 1,2DAG. An intermediate fatty acid that is formed when synthesizing triglycerides for storage.
- TG synthesis generates 1,2DAG
- Accumulation leads to translocation of 1,2DAG into the cell membrane
- 1,2DAG attracts Protein Kinase C (PKC) to the membrane
- PKC internalizes LDLr -> No uptake of cholesterol-rich lipoprotein
- PKC inhibits Insulin Receptor Signaling (IRS) -> insulin resistance!
- 1,2DAG is preferentially hydrolyzed (broken down) for fat metabolism
- Exercise, fasting, high fat diets increase fat metabolism
- Increased clearance of 1,2DAG via DGAT1 expression (exercise)
- Restores insulin sensitivity
- Increases SREBP1 expression -> increases LDLr expression -> increases lipid storage capacity
It does create a conflict though because the accumulation also indicates the need for more cholesterol but 1,2DAG prevents this by blocking LDLr. You cannot signal stop and store more at the same time so how do we end up in this situation?
- Increased storage in lipid droplet reduces cellular cholesterol
- lower cellular cholesterol -> SREBP1 activation -> increase LDLr expression
- 1,2DAG accumulation threshold blocks LDLr via PKC
- Prioritize cellular cholesterol production instead of via extracellular uptake
- Reduce cellular fatty acid accumulation
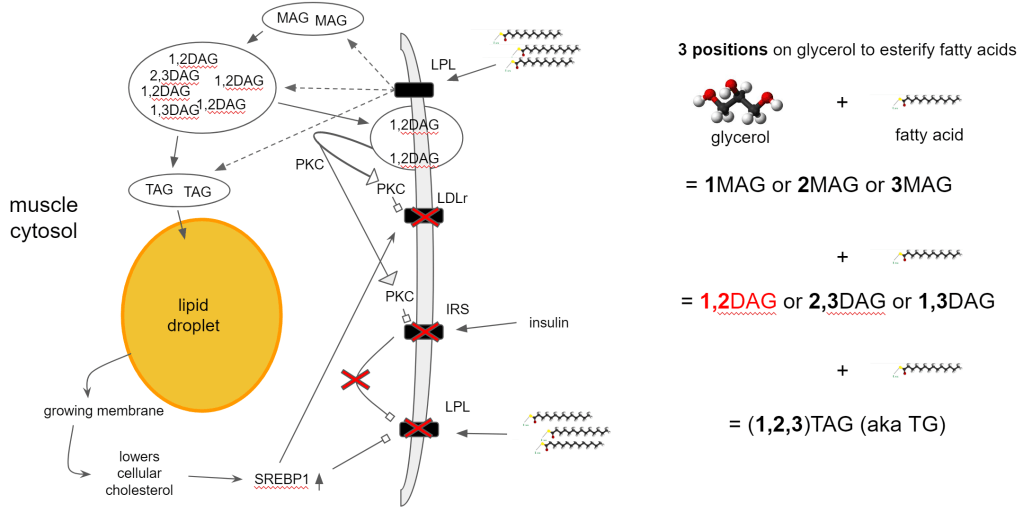
In and of itself this is a normal mechanism. By not taking up both lipids and membrane components, other cells can manage to top up their reserves so you get an even distribution. The problem however is when every cell is topped up and you still have too much in circulation.
It will lead to higher insulin levels because too much energy in circulation drives up insulin to try and store that excess. That will lead to more VLDL-bound TG, higher CETP thus higher cholesterol bound to LDL (higher cholesterol/particle ratio) and lower HDL.
But with cells that have enough fat accumulated, they say no to insulin so insulin does what it is supposed to do, form the lipoprotein particles that are required for storage but all the cells keep their doors closed.
Exactly the pathologic lipid profile that we know and should fear.
- Increased VLDL triglycerides
- Increased cholesterol-rich LDL
- Decreased HDL (lower cholesterol/particle ratio)
Why should we fear it? Because it has a good proxy value towards atherosclerosis.
Atherosclerosis
saved by enhanced fat metabolism?
As mentioned in the introduction, it may actually mean reversal of plaque! The very low insulin obtained through being lean, very low carbohydrate diet with high fat intake creates cholesterol-enriched – ApoE-enriched HDL which drives up their LCAT activity. LCAT stimulates cholesterol release from macrophages.
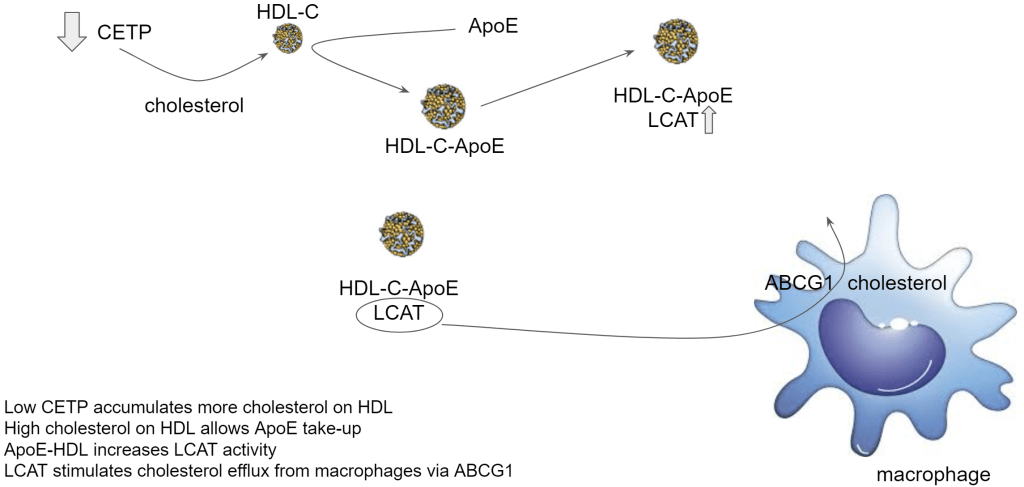
Macrophages in atherosclerosis aren’t only gobbling up cholesterol, they also accumulate triglycerides. It turns out though that cholesterol efflux helps to switch gears into fat metabolism so the macrophage can reduce its fat storage. If you cannot store the fat, you have to start using it for energy.
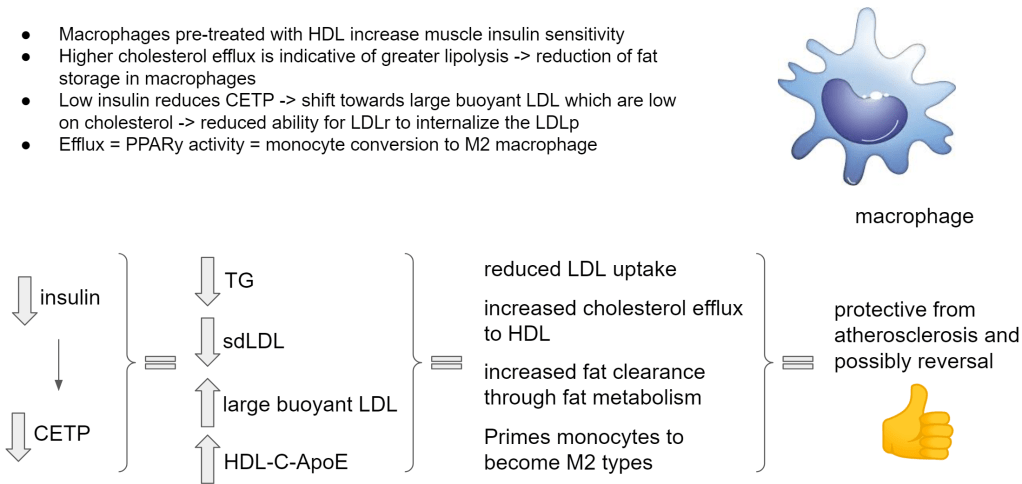
This whole deep dive into research led me to the following, to my view important, observation. These M1 macrophages actually maintain import and storage of fatty acids (through LDLr). Only by ‘forcing’ cholesterol efflux are we able to reverse the situation, pushing them to metabolize the fat. PPARy is known to do this and PPARy is activated by BHB, thus a ketogenic diet. PPARy is also activated under low cellular cholesterol, via SREBP1. This has been evidenced in liver and fat cells but likely also applies to macrophages. And a deactivation of PPARy was accomplished by re-addition of cholesterol.
PPARy expression hence seems subject to a tight and fast control by alterations in intracellular cholesterol levels, and this effect is mediated by the SREBP family of transcription factors.
source: https://pubmed.ncbi.nlm.nih.gov/10409739/ “Regulation of peroxisome proliferator-activated receptor gamma expression by adipocyte differentiation and determination factor 1/sterol regulatory element binding protein 1: implications for adipocyte differentiation and metabolism”

A paper showed me that macrophages are able to restore insulin resistance in skeletal muscle of diabetics. Insulin resistance that is caused by 1,2DAG as we have seen. So if macrophages carry the capacity to somehow neutralize 1,2DAG from the membrane then maybe they apply this capacity also to their own membrane.
This is still an area I need to find research on but we already see that macrophages do not exhibit a stop mechanism like other cells.
That would explain why they get so big with fat and cholesterol. The only way out of this situation is to enhance the efflux of cholesterol from the macrophages. And the only way to do that is by creating big cholesterol-rich HDL so that it can acquire ApoE and drive up LCAT. And the only way to do that is by getting your insulin very low. And the only way to do that sustainably(!) is by adopting a very low carb diet and getting lean.
Get those cells to push out cholesterol like it’s nobodies business!
update 2021/09/21: A paper came out showing how BHB could be reversing vascular calcification through autophagy. Although it is separate from the described mechanism above, it does pertain to enhancing fat metabolism as a way to reverse the diseased state.
“β-Hydroxybutyric Inhibits Vascular Calcification via Autophagy Enhancement in Models Induced by High Phosphate.” https://www.ncbi.nlm.nih.gov/labs/pmc/articles/PMC8422966/
update 2022/09/23: A paper shows us that through HDAC9 downregulation, BHB suppresses vascular calcification. Also different from the described mechanism but just like the previous update it is an extra element supporting potential reversal.
“Downregulation of HDAC9 by the ketone metabolite β-hydroxybutyrate suppresses vascular calcification” https://onlinelibrary.wiley.com/doi/10.1002/path.5992
—– T H E – E N D —–
Sources
My previous writings on this topic with references that over time have contributed to my understanding.
- Insulin resistance
- Oxidized LDL
- Cholesterol or ketones, can we have both?
- LMHR and the elevated LDL cholesterol
- The curious case of macrophages and LDL cholesterol uptake in the pathology of atherosclerosis
- The cause of atherosclerosis is unknown… or is it?
- Pathologic LDL cholesterol
- CETP
- (earliest work) LDL
Further resources, specifically for this article
LXR
https://pubmed.ncbi.nlm.nih.gov/12697904/ “Activation of liver X receptor improves glucose tolerance through coordinate regulation of glucose metabolism in liver and adipose tissue”
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6433546/ “Liver X receptors in lipid signaling and membrane homeostasis”
Cholesterol efflux
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4159082/ “Cholesterol efflux and atheroprotection: Advancing the concept of reverse cholesterol transport”
https://pubmed.ncbi.nlm.nih.gov/17390217/ “Stimulation of lipolysis enhances the rate of cholesterol efflux to HDL in adipocytes”
Lipids on a ketogenic diet
https://academic.oup.com/jn/article/135/6/1339/4663837 “Modification of Lipoproteins by Very Low-Carbohydrate Diets”
LPL receptor
https://www.ncbi.nlm.nih.gov/books/NBK537040/ “Biochemistry, Lipoprotein Lipase”
https://www.sciencedirect.com/science/article/pii/S0022227520380901 “Effects of dietary carbohydrate and fat on plasma lipoproteins and apolipoproteins C-II and C-III in healthy men”
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5705268/ “Apolipoprotein C-II: New findings related to genetics, biochemistry, and role in triglyceride metabolism”
https://www.ahajournals.org/doi/full/10.1161/01.ATV.19.3.472 “Role of ApoCs in Lipoprotein Metabolism”
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5705268/ “Apolipoprotein C-II: New findings related to genetics, biochemistry, and role in triglyceride metabolism”
https://pubmed.ncbi.nlm.nih.gov/11090277/ “Induction of LPL gene expression by sterols is mediated by a sterol regulatory element and is independent of the presence of multiple E boxes”
LDL in skeletal muscle
https://pubmed.ncbi.nlm.nih.gov/9696990/ “Skeletal muscle lipoprotein lipase: molecular regulation and physiological effects in relation to exercise”
https://www.sciencedirect.com/science/article/pii/S0925443902001692 “Expression and regulation by insulin of low-density lipoprotein receptor-related protein mRNA in human skeletal muscle”
https://diabetes.diabetesjournals.org/content/50/11/2585 “In Muscle-Specific Lipoprotein Lipase−Overexpressing Mice, Muscle Triglyceride Content Is Increased Without Inhibition of Insulin-Stimulated Whole-Body and Muscle-Specific Glucose Uptake”
https://pubmed.ncbi.nlm.nih.gov/17046550/ “Fasting decreases free fatty acid turnover in mice overexpressing skeletal muscle lipoprotein lipase”
in liver
https://pubmed.ncbi.nlm.nih.gov/12951168/ “Mechanisms of HDL lowering in insulin resistant, hypertriglyceridemic states: the combined effect of HDL triglyceride enrichment and elevated hepatic lipase activity”
LDL receptor
https://www.ahajournals.org/doi/10.1161/circresaha.114.301621 “PCSK9 – A key modulator of cardiovascular health”
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3071588/ “Activation of LDL Receptor (LDLR) Expression by Small RNAs Complementary to a Noncoding Transcript that Overlaps the LDLR Promoter”
https://pubmed.ncbi.nlm.nih.gov/16449296/ “Exercise training and calorie restriction increase SREBP-1 expression and intramuscular triglyceride in skeletal muscle”
https://diabetes.diabetesjournals.org/content/69/5/848 “Exercise and Muscle Lipid Content, Composition, and Localization: Influence on Muscle Insulin Sensitivity”
https://pubmed.ncbi.nlm.nih.gov/9010277/ “Low density lipoprotein receptor expression and function in human polymorphonuclear leucocytes”
https://pubmed.ncbi.nlm.nih.gov/3237238/ “The binding of lipoproteins to human muscle cells: binding and uptake of LDL, HDL, and alpha-tocopherol”
https://journals.physiology.org/doi/full/10.1152/ajpendo.00543.2005 “Exercise training and calorie restriction increase SREBP-1 expression and intramuscular triglyceride in skeletal muscle”
https://www.cell.com/fulltext/S0092-8674(00)80213-5 “The SREBP Pathway: Regulation of Cholesterol Metabolism by Proteolysis of a Membrane-Bound Transcription Factor”
https://www.ahajournals.org/doi/full/10.1161/01.ATV.18.3.466 “Influence of ApoE Content on Receptor Binding of Large, Buoyant LDL in Subjects With Different LDL Subclass Phenotypes”
https://pubmed.ncbi.nlm.nih.gov/8254047/ -> https://dm5migu4zj3pb.cloudfront.net/manuscripts/116000/116915/cache/116915.1-20201218131521-covered-253bed37ca4c1ab43d105aefdf7b5536.pdf “Accumulation of “small dense” low density lipoproteins (LDL) in a homozygous patients with familial defective apolipoprotein B-100 results from heterogenous interaction of LDL subfractions with the LDL receptor”
https://pubmed.ncbi.nlm.nih.gov/16670767/ “Putting cholesterol in its place: apoE and reverse cholesterol transport”
Lipids
https://www.sciencedirect.com/science/article/pii/S0022227520380901 “Effects of dietary carbohydrate and fat on plasma lipoproteins and apolipoproteins C-II and C-III in healthy men”
https://www.britannica.com/science/lipid/Classification-and-formation “Classification and formation”
Macrophages
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3767414/ “Lipid Droplets And Cellular Lipid Metabolism”
https://journals.plos.org/plosone/article?id=10.1371%2Fjournal.pone.0056601 “Skeletal Muscle Insulin Resistance Associated with Cholesterol-Induced Activation of Macrophages Is Prevented by High Density Lipoprotein”
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1868788/ “Macrophage PPARγ is required for normal skeletal muscle and hepatic insulin sensitivity and full antidiabetic effects of thiazolidinediones”
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3957345/ “High-Density Lipoprotein Maintains Skeletal Muscle Function by Modulating Cellular Respiration in Mice”
https://pubmed.ncbi.nlm.nih.gov/29217413/ “Apolipoprotein E-containing high-density lipoprotein (HDL) modifies the impact of cholesterol-overloaded HDL on incident coronary heart disease risk: A community-based cohort study”
https://pubmed.ncbi.nlm.nih.gov/16670775/ “HDL from CETP-deficient subjects shows enhanced ability to promote cholesterol efflux from macrophages in an apoE- and ABCG1-dependent pathway”
https://www.nature.com/articles/aps201093 “A novel model of cholesterol efflux from lipid-loaded cells”
https://www.cell.com/cell-metabolism/references/S1550-4131(07)00166-0 “PPARγ Activation Primes Human Monocytes into Alternative M2 Macrophages with Anti-inflammatory Properties”
https://pubmed.ncbi.nlm.nih.gov/22207731/ “Regulation of lipid droplet cholesterol efflux from macrophage foam cells”
https://www.nature.com/articles/s41598-018-34305-x “A reduced M1-like/M2-like ratio of macrophages in healthy adipose tissue expansion during SGLT2 inhibition”
Lipid droplet
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4575688/ “DAG tales: the multiple faces of diacylglycerol—stereochemistry, metabolism, and signaling”
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4526153/ “The Biophysics and Cell Biology of Lipid Droplets”
Muscle cell
https://www.sciencedirect.com/science/article/pii/S0005273615003867 “Alteration of lipid membrane structure and dynamics by diacylglycerols with unsaturated chains”
https://journals.plos.org/plosone/article?id=10.1371%2Fjournal.pone.0056601 “Skeletal Muscle Insulin Resistance Associated with Cholesterol-Induced Activation of Macrophages Is Prevented by High Density Lipoprotein”
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1665384/ “High triacylglycerol turnover rate in human skeletal muscle”
https://journals.physiology.org/doi/full/10.1152/ajpendo.00543.2005 “Exercise training and calorie restriction increase SREBP-1 expression and intramuscular triglyceride in skeletal muscle”
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1866250/ “Upregulation of myocellular DGAT1 augments triglyceride synthesis in skeletal muscle and protects against fat-induced insulin resistance”
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3510842/ “Studies on the Substrate and Stereo/Regioselectivity of Adipose Triglyceride Lipase, Hormone-sensitive Lipase, and Diacylglycerol-O-acyltransferases”
Dawn phenomenon
https://joe.bioscientifica.com/view/journals/joe/231/3/235.xml “Melatonin modifies basal and stimulated insulin secretion via NADPH oxidase”
https://pubmed.ncbi.nlm.nih.gov/6368151/ “Fasting early morning rise in peripheral insulin: evidence of the dawn phenomenon in nondiabetes”
PPARy
https://pubmed.ncbi.nlm.nih.gov/10409739/ “Regulation of peroxisome proliferator-activated receptor gamma expression by adipocyte differentiation and determination factor 1/sterol regulatory element binding protein 1: implications for adipocyte differentiation and metabolism”

Leave a comment