Read it carefully as it may help you assess your risk for cardiovascular disease!
In my previous article comparing pathological versus physiological high LDL-C levels, one of the marked differences is that HDL-C goes down under the pathological situation versus up on the physiological case.
This difference comes down to cholesterol ester transfer protein or CETP. That has triggered my curiosity so naturally I wanted to understand more of it.
What does it do? Where does it come from? And since we see our HDL-C levels modulated by diet due to this CETP activity.. how does diet modulate CETP?
Exchanger
First, what does it do? CETP is responsible for the exchange of triglycerides for cholesterol between the lipid particles. HDL-particles will give up a cholesterol ester in exchange for a triglyceride from ApoB-containing particles such as VLDL, IDL or LDL.
What normally happens is that the HDL-particle gets loaded up with triglycerides. The liver will take up triglyceride rich HDL-particles via hepatic lipase. Stripped from its triglycerides, the remnant HDL is put back into circulation but risks higher clearance via the kidneys.
“New insights into the mechanism of low high-density lipoprotein cholesterol in obesity – Changes in HDL component” https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3207906/#__sec6title
This explains why we see HDL-C trend upwards for athletes and lean individuals on a ketogenic diet. It is due to their CETP activity being reduced.
But it raises a question. It could explain why LDL particles become large buoyant in size as the particle is not unloading its triglycerides to the HDL particle. But the HDL particle normally also exchanges with VLDL particles so how come the lipid profile results in low triglyceride levels under reduced CETP activity?
This may be difficult to answer but could be driven by a reduction in VLDL-sized ApoB particles. Under fasted conditions, the liver may be producing LDL-sized ApoB particles directly which is true under prolonged fasted conditions where the liver is more tuned towards fat metabolism.
Macrophages
Low CETP activity induces higher cholesterol efflux from macrophages to HDL-particles. When we look at subjects with homozygous CETP deficiency we see a 2~3-fold increase in efflux.
“HDL from CETP-deficient subjects shows enhanced ability to promote cholesterol efflux from macrophages in an apoE- and ABCG1-dependent pathway” https://pubmed.ncbi.nlm.nih.gov/16670775/
But we are not all genetically deficient in CETP. When using a drug that inhibits CETP activity, we also not a higher efflux capacity.
“Cholesterol Efflux Potential and Anti-inflammatory Properties of HDL following Treatment with Niacin or Anacetrapib” https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2917780/
Metabolic syndrome
Given that lower CETP activity is beneficial as it increases HDL-C and causes a greater efflux of cholesterol from macrophages, we want to know what the level is for ourselves.
HDL-C by itself is already a good proxy but the following study shows that CETP activity is also associated with increased BMI, fasting glucose and c-peptide. This explains why obese people tend to have lower HDL-C
“Effect of adiposity on plasma lipid transfer protein activities: a possible link between insulin resistance and high density lipoprotein metabolism” https://pubmed.ncbi.nlm.nih.gov/8033953/
When reviewing patients with metabolic syndrome, they found increased CETP activity (and PCSK9).
“Circulating PCSK9 levels and CETP plasma activity are independently associated in patients with metabolic diseases” https://cardiab.biomedcentral.com/articles/10.1186/s12933-016-0428-z
Small dense LDL
In type 2 diabetes patients it has been demonstrated that CETP contributes significantly to the increased levels of small dense LDL by preferential CE transfer from HDL to small dense LDL, as well as through an indirect mechanism involving enhanced CE transfer from HDL to VLDL-1(114). … Recent evidence has indicated that a primary acceptor for CETP-mediated HDL cholesteryl ester transfer in normolipidemic subjects is a large, buoyant, triglyceride-enriched LDL subclass (116).
source: “Metabolic origins and clinical significance of LDL heterogeneity” https://www.sciencedirect.com/science/article/pii/S0022227520328005
CETP targets the large buoyant LDL particles and turns them into small dense LDL particles. Small dense LDL is a biomarker for atherosclerosis.
Here are some numbers from a study looking at events across 2 years in people with metabolic syndrome. In those with events: LDL size was lower (P < 0.0001), due to reduced larger subclasses and increased small, dense LDL (all P < 0.0001). After multivariate analysis for independent risk factors: elevated small, dense LDL (OR 11.7, P = 0.0004). Now that is impressive.
“Small Dense Low-Density Lipoprotein as Biomarker for Atherosclerotic Diseases” https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5441126/
“Small, dense low-density lipoproteins (LDL) are predictors of cardio- and cerebro-vascular events in subjects with the metabolic syndrome” https://pubmed.ncbi.nlm.nih.gov/18771560/
Insight
If you paid close attention to the above then you may have noticed something.
HDL-particles have 2 jobs. Doing one makes it bad at the other and this is regulated by CETP activity.
- Recycling of triglycerides from LDL-particles to the liver
- Taking up cholesterol from macrophages
When CETP activity is high, HDL volume reduces from circulation because it is taken up by the liver (and also by the kidneys). With less of them available, there is less availability to take up cholesterol from macrophages.
When CETP is low, there is greater capacity to take up cholesterol from macrophages. As a result LDL particles will have to hold on to their triglycerides more.
CETP activator
So what causes CETP to be activated or turned down?
CETP is secreted by the liver and this is regulated by liver x protein (LXP). Higher stimulation of LXP causes higher levels of CETP.
Interestingly higher LXP also causes a higher stimulation of GLUT5 expression in the duodenum and adipose tissue.
“Identification of the fructose transporter GLUT5 (SLC2A5) as a novel target of nuclear receptor LXR” https://www.nature.com/articles/s41598-019-45803-x
The duodenum is particularly interesting because it is thought that most of the fructose is processed by the bacteria in the small intestine. This idea is based on the following study which, to my view, people incorrectly extrapolate to humans. The study was done in mice and I doubt they have a similar gut microbiome. Secondly fructose that would be absorbed by the liver and converted to fat would not immediately show up in the circulation.
“The Small Intestine Converts Dietary Fructose into Glucose and Organic Acids” https://www.sciencedirect.com/science/article/pii/S1550413117307295
It is important to find out what causes higher expression of LXP. Causing more fructose to be absorbed into the body is really not a good thing. It already creates a bad lipid profile for cardiovascular disease.
LXP activity
I figured it had something to do with fat metabolism since we see evidence of very low CETP activity in very lean individuals on a ketogenic diet. The diet features things such as PPAR-alpha increase, PGC-1alpha increase, AMPK increase. So a quick look around and indeed, it seems that AMPK activations directly inhibits liver x receptor (LXR). The receptor is a protein and sometimes referred to as LXP.
These results indicate that AMPK directly inhibits ligand-induced LXR activity in addition to blocking production of endogenous LXR ligands.
source: “Mechanism of AMPK suppression of LXR-dependent Srebp-1c transcription” https://pubmed.ncbi.nlm.nih.gov/21647332/
This indicates that lean ketogenic individuals have remarkably elevated AMPK levels. It shouldn’t be a surprise really. This has already been studied very well. An example here shows that AMPK levels double in the liver when the animals are put on a ketogenic diet. 2-fold higher in the liver and 2 to 3-fold higher in muscle.
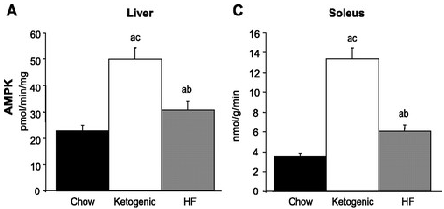
“A high-fat, ketogenic diet induces a unique metabolic state in mice” https://journals.physiology.org/doi/full/10.1152/ajpendo.00717.2006
Pharma Failure?
It is tempting that given all of the above you can simply develop a drug to inhibit CETP. So they did and had to stop the trial early because it caused more deaths. I couldn’t find much input about due to what the patients died from but there was a follow-up study that indicates it was not caused by the inhibition of CETP but due to side-toxicity.
The study said the following about the CETP inhibition:
Recent analysis suggests that failure may have been caused by off-target toxicity and that HDL is functional and promotes regression of atherosclerosis. New studies highlight the central importance of the ATP-binding cassette (ABC) transporters ABCA1 and ABCG1 in reducing macrophage foam cell formation, inflammation, and atherosclerosis. A variety of approaches to increasing HDL may eventually be successful in treating atherosclerosis.
source: “HDL, ABC transporters, and cholesterol efflux: implications for the treatment of atherosclerosis” https://pubmed.ncbi.nlm.nih.gov/18460328/
Finally
It seems clear to me that you need to get into a state where your body lowers CETP activity naturally and thereby increases your HDL-C. In my previous article on pathological high LDL we clearly see that a low HDL-C is a risk indicator along with obesity etc.
My advice: get onto the ketogenic diet, get lean and smile when you see your HDL-C rising because you know it is working.
I wish you Good Luck and enjoy the rest of your day!
—- T H E – E N D —-

Leave a comment