One of the things I noticed in studying metabolism and the overall functioning of the body, is how important the liver is. A lot of what we do and a lot of what we measure is influenced by the liver and its buffers. Understanding these buffers in conjunction with these measurements may help us a great deal in evaluating health and disease.
There is a lot of controversy on what we measure and if that is healthy or not. I think most of this controversy is created due to lack of context. As we all acknowledge, an obese Type 2 diabetic (T2D) person is vastly different from a lean athlete and numbers measured have to be interpreted accordingly. Someone on a ketogenic diet with high ketone levels (>1mmol/L) can walk around just fine with 50mg/dL glucose while someone else on a Standard American Diet (SAD) will experience hypoglycemia at 65mg/dL glucose.
It is the same with diet, a low fat high carb (LFHC) diet will stimulate hormones differently than a high fat low carb (HFLC) diet. And that difference is also there when comparing a diet high in processed foods versus whole foods etc.
My goal is to provide that context to provide you with a better understanding. Apart from sugar I will not get into processed food. Processed food is a big failure in our human history.
What are the elements that we will look at?
There are 2 main hormones (insulin and glucagon) which influence the buffers in the liver so we’ll look at what changes those hormones in various situations such as food, exercise, fasting etc.
I’ll explain what happens over time according to my current understanding. As I’m self-educated I appreciate any input and certainly to correct any misrepresentations.
We’ll have a look at how insulin and glucagon levels are modified but also the functions they stimulate or block in the liver. We’ll look at the following aspects:
- Glucagon
- gluconeogenesis (GNG): creating glucose from substrates such as lactate, glycerol, amino acids
- glycolysis: breakdown of glycogen to form glucose
- lipolysis: breakdown of fat (triglycerides or triacylglycerol or TAG) into free fatty acids so not bound to their glycerol backbone
- ketogenesis: creating ketones, we’ll talk about beta-hydroxybutyrate (BHB) specifically
- Insulin
- glycogenesis: converting glucose into glycogen
- cholesterol: cholesterol production through ApoB100 lipoprotein (VLDL, IDL, LDL) efflux from the liver. I’ll be mentioning ApoB in the article but talking about ApoB100 specifically.
- de novo lipogenesis (DNL): creating fat from glucose
- de novo lipogenesis (DNL): creating fat from fructose
I listed DNL twice. That is because DNL from fructose is done without insulin playing a role. This makes an interesting combination in sugar. I will get more into it in the sugar situation below.
“De novo lipogenesis in the liver in health and disease: more than just a shunting yard for glucose” https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4832395/
The buffers that we will look at are those of the glycogen (stored glucose) and lipids. A few words may be at place for why I think those buffers exist. This may help understanding the changes that we see taking place in the scenarios below.
Glycogen
This is our glucose reserve. The body strives to maintain a constant level of blood glucose at rest. Under different situation, it may deviate from this homeostatic level so a buffer is needed to respond to these changes. Below are a few examples on how blood glucose is regulated.
Elevate: Viral infections need to be countered with elevated glucose levels to stimulate our immune cells to proliferate. Exercise can rapidly consume glucose so more glucose must be released in order to maintain the right level.
Maintain: The brain is an energy hungry animal frequently quoted as being 2% of our body weight while consuming 20% of the energy. It requires a steady influx of energy all the time.
Suppress: During times of food deprivation we need to switch towards preferring fat so that we can spare glucose or otherwise the body will increasingly target protein sources for GNG. More on that under the fasting scenario.
Lipids
The liver is also metabolically active. Depending on the situation it may be consuming more glucose or more fatty acids. Either way and no matter how it got a hold of the fat, when allowed to, it will release and clear itself of the temporarily stored fat to redistribute it across the body.
Contrary to glycogen, the liver does not try to maintain a lipid buffer. It temporarily holds lipids to enhance absorption and redistributes the lipids afterwards. It is like our adipose tissue but more of a temporary buffer that will be consumed first before switching to adipose (in a simplified way).
So let’s start with the fasted state, probably the most well known state with the least controversy.
Fasting
With fasting I mean prolonged fasting. Not the time between for example lunch and dinner and also not the time between meals such as in One Meal A Day (OMAD) so multiple days without food.
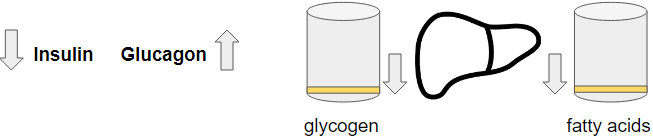
GNG – Under fasting, both buffers will go down. There is no stimulation of insulin so they will drop down to their lowest level. That allows glucagon to dominate. Because glycogen levels go down, the contribution of glycogen to the blood glucose goes down as well. This means that GNG will become the highest contributor to blood glucose. So much that after 40 hours we can see GNG delivering 96% of the circulating glucose.
“Quantitation of Hepatic Glycogenolysis and Gluconeogenesis in Fasting Humans With 13C NMR”, https://science.sciencemag.org/content/254/5031/573
Lactate – With a buffer going empty, what are the substrates for GNG? At first there may be some lactate still but lactate will go down. Glucose will go down over time and is the substrate in metabolism that will generate the lactate. Glucose goes down because there is not sufficient substrate available to keep up the homeostatic level through GNG.
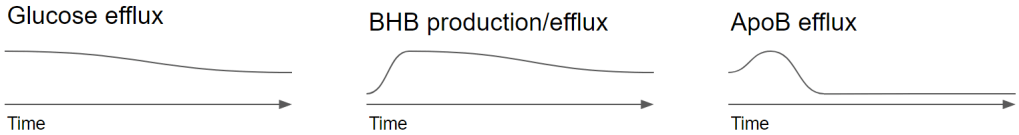
BHB – Depending on our available fat mass, BHB will be produced in sufficient quantity to make up for the lack of glucose. The brain is equally happy with BHB as it is with glucose. And at the same time, the glycerol from the fatty acid breakdown provides a source for GNG. But at a certain point, our fat mass will reduce too much and become insufficient to support the right level of BHB production leading to a declining BHB level and that will also be paralleled with a decline in glycerol.
Protein – If neither glucose nor BHB levels can be maintained sufficiently for the brain, protein will start to be broken down more and more to be a source for GNG. The whole system works in such a way that it tries to spare protein until it is no longer possible. The brain must continue to function and continues to consume energy.
The liver glycogen level is an important buffer for glucose but as it runs out, BHB will provide protection from protein breakdown. Fasting cannot be maintained forever (although the record of 382 days is quite long). The fat will eventually be insufficient as you get lean so gradually protein breakdown will go up to continue sourcing energy for the brain.
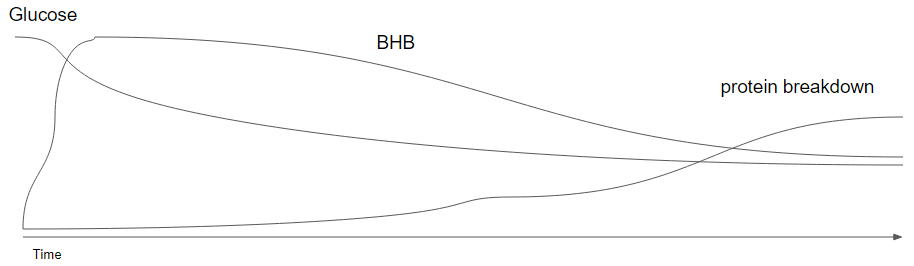
Ignore the bumps in the lines below, I’m a lousy drawer 😉 Also ignore the proportions. The general concept is that at the beginning glucose goes down quickly and then over a longer period gradually goes down further. BHB goes up sharply over the first few days and then over a longer period gradually will go down. Protein breakdown will gradually go up over a long period as glucose and BHB become insufficient.
But the body is clever with protein, notice there is no excess skin in the face of our 382 days record holder (check the picture via the link). Also if you look at pictures from the unfortunate concentration camp prisoners during the world war, they have no excess skin.
Cholesterol – From the moment we start fasting the liver releases the stored triglyceride. This will create an abundance of acetyl-coa so that temporarily ApoB lipoprotein output can increase. ApoB efflux increases with dropping insulin but only when it has sufficient material to load such as cholesterol and lipids. Otherwise ApoB breaks down again. The increase in glucagon will also divert those acetyl-coa towards ketone production. This means that ApoB will quickly drop because the liver can’t maintain cholesterol production due to glucagon and with the lipid buffer being cleared up, we don’t have an abundance of acetyl-coa so the majority will go out the liver as a ketone.
Fat – With the liver being cleared from its lipid storage, the fatty acids it can obtain are now coming from the circulation. This is the reason why BHB will drop over time. Our fat stores will shrink with less and less fatty acids reaching the liver.
Sugar (glucose & fructose)
Let’s review now what happens if we take in sugar or high fructose corn sirup (HFCS), both will give us a roughly equal intake of glucose and fructose. Notice the 2 arrows for glucagon, explanation follows.

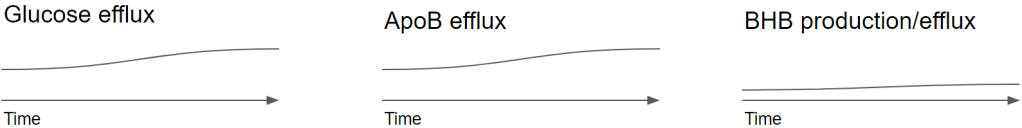
Glucose – A first thing we note is that both buffers are increased. High glucose flowing in the body provides a great stimulation for insulin secretion. This will suppress glucagon release and we don’t have any other elements that would stimulate glucagon secretion.
High glucose availability under high levels of insulin will convert glucose to fatty acids. Insulin stimulates glycogenesis so glucose causes both an increase in glycogen buffer and in fatty acids but limited to the period in which insulin is elevated.
Fructose – Now keep in mind we also have fructose as part of the sugar (half glucose, half fructose). The fructose that gets absorbed into the body will be fully converted to fatty acids in the liver, increasing the lipid buffer. How much gets absorbed depends on the volume, speed and frequency of how you take in the fructose. Sugar sweetened beverages are well associated with non-alcoholic fatty liver disease (NAFLD)
“Consumption of Sugar-Sweetened Beverages Has a Dose-Dependent Effect on the Risk of Non-Alcoholic Fatty Liver Disease: An Updated Systematic Review and Dose-Response Meta-Analysis” https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6617076/
What is remarkable is that frequent, fast and large enough quantities of fructose will create hepatic insulin resistance. If this influx of fructose is repeated often enough over time then the signaling of insulin to the liver starts to fail leading to more and more fatty acids export.
At some point the adipose fat cells are full and those fatty acids start to accumulate as visceral fat in and around the organs.
The pancreas is an important organ in this case because the visceral fat will make it also less and less responsive to insulin with a reduction of control on the glucagon secretion. Due to the hepatic insulin resistance and increasing glucagon, glycogenesis will reduce, GNG will increase and glycogenolysis will increase leading to higher plasma glucose and certainly high glucose spikes with subsequent sugar intake.
This is why there are 2 arrows next to glucagon. At first the increasing insulin will lower glucagon when the pancreas is still responsive but as we progress over time, insulin fails to affect glucagon secretion due to the visceral fat in the pancreas so that glucagon goes up as insulin looses its grip on the production.
ApoB – Post-prandially the liver secretion of ApoB particles will become high as insulin subsides. Because we had high insulin, cholesterol production went up, we have increased the lipids and insulin also breaks down ApoB. With insulin going down we increase ApoB production again and now have abundant cholesterol and lipids for ApoB secretion from the liver.
Cholesterol – As we build up insulin resistance over time, the ApoB secretion will increase. Cholesterol production depends on insulin signaling and is a necessary component for secretion but lipid availability is far more important. So why do we see elevated cholesterol levels? As insulin resistance builds up, the LDL receptors (LDLr) will go down which normally take out ApoB lipoprotein from the circulation. The ApoB output will be higher than the clearance resulting in a buildup.
In addition, what does get secreted doesn’t find a place anymore. With adipose tissue full, not accepting any more lipids and visceral fat already building up, the triglycerides remain in circulation longer. The lipid catabolism of the ApoB particles run at a slower rate. The result is elevating cholesterol and triglycerides.
“Apo B secretion is regulated by hepatic triglyceride, and not insulin, in a model of increased hepatic insulin signaling” https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3870321/
“The Regulation of ApoB Metabolism by Insulin” https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3810413/
BHB – I’m missing some clear data but I suspect that over time, if this situation continues then BHB production will go up slightly due to glucagon secretion increasing and liver response to insulin decreasing. With glucose available in abundance, probably acetyl-coa will become available in abundance in the liver mitochondria allowing some to escape into the ketogenic pathway. This situation is also recognized in the literature by case reports of ketoacidosis in T2D patients. Below is a review referenced.
“Update on diagnosis, pathogenesis and management of ketosis-prone Type 2 diabetes mellitus” https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3351851/
As the situation gets worse and worse, also the insulin secretion by the pancreas may become impaired. This leads to further increase of glucose release from the liver. You could think with all this glucose output and VLDL output that the liver becomes depleted but keep in mind we are talking about continuous repeated intake of sugar. This is a situation that builds up over several years and is now a problem that even our children are affected with.
“Non-Alcoholic Fatty Liver Disease in Overweight Children: Role of Fructose Intake and Dietary Pattern” https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6165138/
The system works out in such a way that as the liver reaches the maximum or is at the maximum of its buffer and there is an overflow to the rest of the organs, glucagon is increasingly elevated and insulin, although increased at first due to the insulin resistance, will go down. This allows a maximum clearance of the liver but by continuing the sugar intake, it will continue to fail.
Notice how I covered the risk factors (obesity, insulin resistance, hypertriglyceridaemia) in the reference below. Yet the driving force sugar,as explained in this scenario, is not mentioned.
“Fatty liver disease in children” https://adc.bmj.com/content/89/7/648
Exercise
What happens during and post exercise? There is a lot going on but I’ll try and stick close to the liver as much as possible.
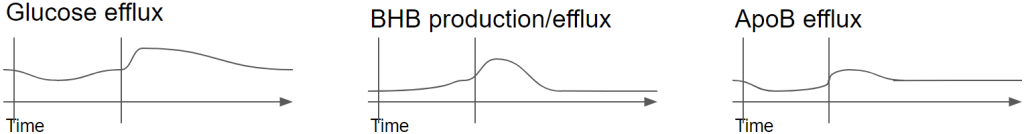
We see that exercise is somewhat similar to fasting but the difference in activity causes different reactions.
Glucose – First of all exercise will increase the consumption of glucose leading to an initial drop of plasma glucose. This will lead to a lowering of insulin and an increase in glucagon. Unlike fasting across multiple days, we exercise for only a few hours. For most people this means we start will a decently filled glycogen buffer so we start in a situation where glucose homeostasis can be maintained for a while. This allows to bring up the glucose level as the hepatic glucose output is increased in response to the exercise.
From the moment we stop exercising, glucose levels will shoot up for a while above homeostatic level. This will be corrected in the next minutes to a few hours by a slight increase in insulin so that glucagon goes down and hepatic release of glucose goes down.
“Glucose Metabolism During Exercise in Man: The Role of Insulin and Glucagon in the Regulation of Hepatic Glucose Production and Gluconeogenesis” https://pubmed.ncbi.nlm.nih.gov/9101062/
BHB – During exercise fatty acids will be increasingly released from adipose tissue but this will be picked up directly by albumin and delivered to the muscles. Blood flow to the liver is reduced so we don’t see much fluctuation in BHB. It will depend on the duration and intensity. As we progress in our endurance activity, more fat will be released so there is a chance of small increase in BHB during the activity.
But certainly, from the moment we stop exercising, just like with glucose, the fatty acids release from adipose will go down only gradually so we are in a situation where blood flow to the liver has been restored, the blood is more saturated with fatty acids and glucose levels in the liver went down. We have insulin lowered and glucagon increased. An ideal situation to produce ketones so we see levels of BHB go up right after exercise.
Cholesterol – Here I’m in doubt because most research looks at the prolonged effect of exercise on lipids or the before and after effect but I want to know during exercise itself.
I would need to find tracer studies but lets go by the mechanisms that we know. Insulin lower and glucagon higher. This promotes higher ApoB production but ApoB export depends on the ability to assemble cholesterol and fatty acids onto it. I suspect that the cholesterol production is down during exercise thus VLDL and LDL output may go down.
It really depends on the state of the buffers. If the lipid level in the liver is sufficiently high then some cholesterol synthesis may be going on. On the other hand, if the lipid buffer is fairly low, the total volume of available lipids could be low. The main source of lipids will be from the circulation. An obese person is more likely to have higher liver fat and adipose fat while a lean person is more likely to have low liver fat and low adipose fat.
For the lean person it will rather follow the path of ketogenesis while for the obese person it will be shifted more to cholesterol synthesis but at a reduced rate depending on the state of insulin resistance and liver glycogen level. As I said, context matters 😉
We see that in the study from Jeff Volek, et al. where, over time during the exercise, BHB production goes up. Both groups are fat adapted but the LCD group has lower glycogen levels due to the diet and a higher lipolysis rate. Note that these athletes received a recovery drink at the end of their run so the graphs will be different from mine. Context again.

“Metabolic characteristics of keto-adapted ultra-endurance runners” https://www.sciencedirect.com/science/article/pii/S0026049515003340
Diet
For the diet I want to present 2 relatively extreme sides. We have the currently general recommended a High Carb Low Fat diet (HCLF), and a High Fat Zero Carb diet (HFZC) such as the ketogenic diet. The ketogenic diet does allow for some carbs but for simplicity I’ll consider the situation of zero carbs.
HCLF
I won’t consider fructose intake here. Most of the fructose comes in the form of sugar and as stated at the beginning, I don’t consider processed food. Diet should at least be whole food and 1 or 2 pieces of fruit will not have the same impact as pure sugar.
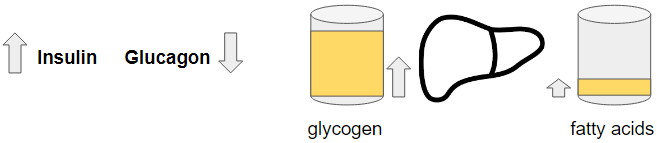

Glucose – With fructose mostly excluded, a high carb diet will primarily bring in glucose as energy so we get an increase as the meal gets digested. As we’ve seen under the sugar scenario, this triggers insulin resulting in increase of glycogen and a smaller part ends up as saturated fatty acids (palmitic acid). How much will depend on the volume of glucose and level of insulin.
Insulin resistance will not establish itself unless perhaps if we start overfeeding on starch rich food on regular basis. This may be induced in part by the dip in glucose levels as insulin starts to push down glucose. After the food intake all other processes will proceed as normal.
Blood glucose levels will be maintained.
BHB – BHB remains suppressed because insulin brought down glucagon, lipolysis and upped the glycogen buffer. Post-absorptive, insulin goes down again restoring glucagon and lipolysis but the glycogen buffer now provides glucose into the liver mitochondria causing the TCA cycle to run at a high enough speed so that there is no excess acetyl-coa for ketone production.
Because the glycogen buffer is full enough, insulin will not go to its lowest level to keep glucagon under control so that there is not too much glucose released from the liver. As such, fasting insulin levels are in function of how well glucagon needs to be suppressed to prevent too much glucose release from the liver.
“The Rapid Changes of Hepatic Glycolytic Enzymes and fructose-1,6-diphosphatase Activities After Intravenous Glucagon in Humans” https://pubmed.ncbi.nlm.nih.gov/4357616/
Cholesterol – Insulin breaks down ApoB so we get a build-up of fatty acids in the liver and an increase in cholesterol production. After a while, as insulin subsides, ApoB production goes up, which will start to clear the temporarily stored fatty acids.
Due to the increase in insulin there was also a stimulation in LDL receptors. Due to the high carb diet, the LDLr are already numerous so that the clearance rate is much higher. This allows for lower LDL values when measuring fasting in the morning.
HFZC
We’ll assume a long term HFZC, not just one meal after a long period of HCLF. With carbs out of the meal we only have fat and protein.
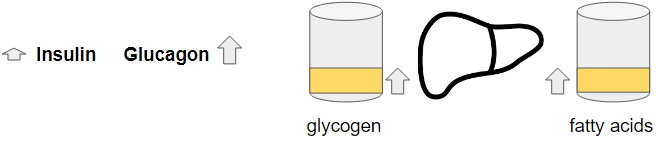

In the fasted state before the meal, because we have a very low glycogen buffer, insulin will be very close to its lowest value possible in order to give glucagon all the power to release the glycogen that remains and stimulate GNG, trying to maintain glucose homeostasis.
Because the meal is missing carbohydrates, we’ll have only a small stimulation of insulin. Glucagon will remain high and maybe increases a bit thanks to the glucagon stimulating amino acids in the protein. There are also insulin stimulating amino acids. Due to the high fat content of the meal, the amino acids will come in at a slower rate. This will also cause a lower stimulation of both hormones so that there is a lower change in their levels.
The high amount of fat will be distributed, some in the adipose, some directly used as fuel throughout the body but also a part is taken up by the liver for later release.
Glucose – Glycogen phosphorylase (breaks down glycogen into glucose) is stimulated by glucagon and inactivated by insulin and separately glycogen synthase (creates glycogen from glucose) is stimulated by insulin. By preventing glycogen breakdown and instead stimulating glycogen creation, the only source for glucose output by the liver will be the GNG while already part of that glucose from GNG will be used for the glycogenesis. This means the glucose output from the liver goes down which would normally cause a drop in blood glucose.
“Mechanism by which glucose and insulin inhibit net hepatic glycogenolysis in humans.” https://www.ncbi.nlm.nih.gov/pmc/articles/PMC508673/
“Activation of Protein Kinase and Glycogen Phosphorylase in Isolated Rat Liver Cells by Glucagon and Catecholamines” https://pubmed.ncbi.nlm.nih.gov/188818/
Such an effect would result in hypoglycemia if it were not for the kidneys. They increase their GNG capacity under the same stimulation of glucagon aiding in the maintenance of homeostatic blood glucose levels.
BHB – The actions of insulin cause a mild buildup of glycogen and a reduction in lipolysis. This will create a temporary reduction in ketone production. As insulin reduces post-absorption, the high fat from the meal is still circulating and lipolysis picks up again creates sufficient supply to the liver to generate ketones.
Cholesterol – ApoB production is already at a very low level before the meal because the liver is as good as fully cleared from fatty acids. What comes in from the blood stream is either metabolized or used for ketone production. Very little will lead to cholesterol production. With the slight increase in insulin, ApoB production will be further prevented so more lipids are stored and cholesterol production is temporarily increased.
Insulin goes down faster after the meal compared to the high carb meal, so we get a quicker increase in ApoB production again. The fatty acids will be cleared and gradually output goes down again.
Regarding the LDLr, before the meal we are at a very low level thanks to the very low level of insulin. The meal will not provide sufficient stimulus to greatly increase the number of LDLr’s. As a result, the temporary increase in ApoB production will lead to higher levels of LDL-c while the clearance remains low.
Bonus info
Under normal circumstances it seems that a full liver glycogen may reduce appetite.
“Liver Glycogen Reduces Food Intake and Attenuates Obesity in a High-Fat Diet–Fed Mouse Model”, Iliana López-Soldado, Delia Zafra, Jordi Duran, Anna Adrover, Joaquim Calbó, and Joan J. Guinovart, 2015, https://diabetes.diabetesjournals.org/content/diabetes/64/3/796.full.pdf
We know the liver and skeletal muscles build up a glycogen reserve but other cells do this as well such as astrocytes in the brain, regulated by Protein Targeting to Glycogen (PTG), and lymphocytes.
“Protein targeting to glycogen is a master regulator of glycogen synthesis in astrocytes” https://www.sciencedirect.com/science/article/pii/S2451830116300152
“The Effect of Immune Cell Activation on Glycogen Storage in the Context of a Nutrient Rich Microenvironment” https://spectrum.library.concordia.ca/980786/
The following paper is remarkably clear in their words. You should not have a full liver glycogen buffer!
“An intermittent exhaustion of the pool of glycogen in the human organism as a simple universal health promoting mechanism” https://www.sciencedirect.com/science/article/abs/pii/S0306987714000206
No matter how high or low the glycogen buffer is, insulin has a steady proportionate influence on hepatic glycogen breakdown and GNG. I’m sure there is a limit to this contribution though. As we’ve seen under fasting, glycogen breakdown can not continue forever and GNG will take the upper hand so the relative contribution shifts.
“Effects of Hepatic Glycogen Content on Hepatic Insulin Action in Humans: Alteration in the Relative Contributions of Glycogenolysis and Gluconeogenesis to Endogenous Glucose Production” https://academic.oup.com/jcem/article/82/6/1828/2656545
— THE END —

Leave a comment