-
Hepatic Glucose Metabolism
After writing about the liver buffers I wanted to understand a bit more on the regulation of gluconeogenesis and buildup of the resulting glucose as glycogen in the liver. I have also written about protein being a supply-driven process with the mechanism intended to increase liver glycogen storage. In order for that supply-driven mechanism to…
-
Linking the hepatic glycogen buffer with protein protection
As a short recap of my article on the liver buffer, insulin causes the build-up of glycogen in the liver. When I looked into protein and fructose, I touched the topic of protein protection for the first time. With this article I wanted to go a bit deeper into this aspect and do this by…
-
LMHR and the elevated LDL cholesterol
For those who don’t know, LMHR stands for Lean Mass Hyper Responder and refers to the lipid profile of people who achieve high LDL cholesterol, high HDL cholesterol and low triglycerides. Hyper responder refers to the high LDL that is achieved and lean mass because most of these people appear to have low body fat.…
-
Longevity (2a)
In the introduction on vaccination there were a couple of questions raised to delve deeper into. In this post we’ll have a look at the following question: Do vaccines get tested for safety in the same (rigorous) way as other drugs? So we need to look at how drugs are tested in general and then…
-
The liver buffer(s)
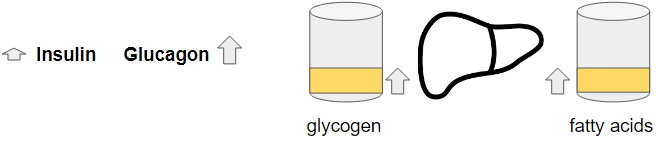
One of the things I noticed in studying metabolism and the overall functioning of the body, is how important the liver is. A lot of what we do and a lot of what we measure is influenced by the liver and its buffers. Understanding these buffers in conjunction with these measurements may help us a…
-
The life-shortening effect of high-protein
Diets come in many forms and some are keen on high protein amounts. While I agree that can be tasty, I’ve come across some material that may put a different light on what that intake means to longevity. There are many aspects to protein and the different type of sources but I’m more interested in…
-
Insulin Resistance
Research in the past and present has shown us that fat causes insulin resistance. With ketogenic diets becoming increasingly popular, many people fear for a potential negative effect by inducing this insulin resistance. It is true that insulin resistance does appear on low carb high fat diets such as the ketogenic diet but the context…
-
Low ketone levels so I utilize them better?
One of the pervasive thoughts in the keto community is that when they measure lower levels of ketones, the longer they are on the ketogenic diet, that this is caused by a better utilization of the ketones in the body. This is an effect noticed over several months or even years. But is this true?…
-
Immune cell modulation by the ketogenic diet
What follows is a list of research that shows how a ketogenic diet can have effect on the immune system. A lot of this research is done in non-human bodies or circumstances so none of it has been proven to be similar in humans. This is very important to know because mice and rats have…
-
Berberine on low carb
I’ve recently came across 2 cases of people who reported elevated glucose and insulin on a low carb diet. Naturally that is alarming because if there is one thing that low carb does well for you, it is to keep glucose and insulin low. It started with a person who recently came to the r/ketoscience…
-
Subscribe
Subscribed
Already have a WordPress.com account? Log in now.
